
Performance Characteristics of Oncomine Focus Assay for Theranostic Analysis of Solid Tumors, A (21-Months) Real-Life Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Section
2.2. DNA and RNA Extraction
2.3. Library Preparation and Sequencing
2.4. Bioinformatic Pipeline
2.5. Immunohistochemistry
3. Results
3.1. Sequencing Performances
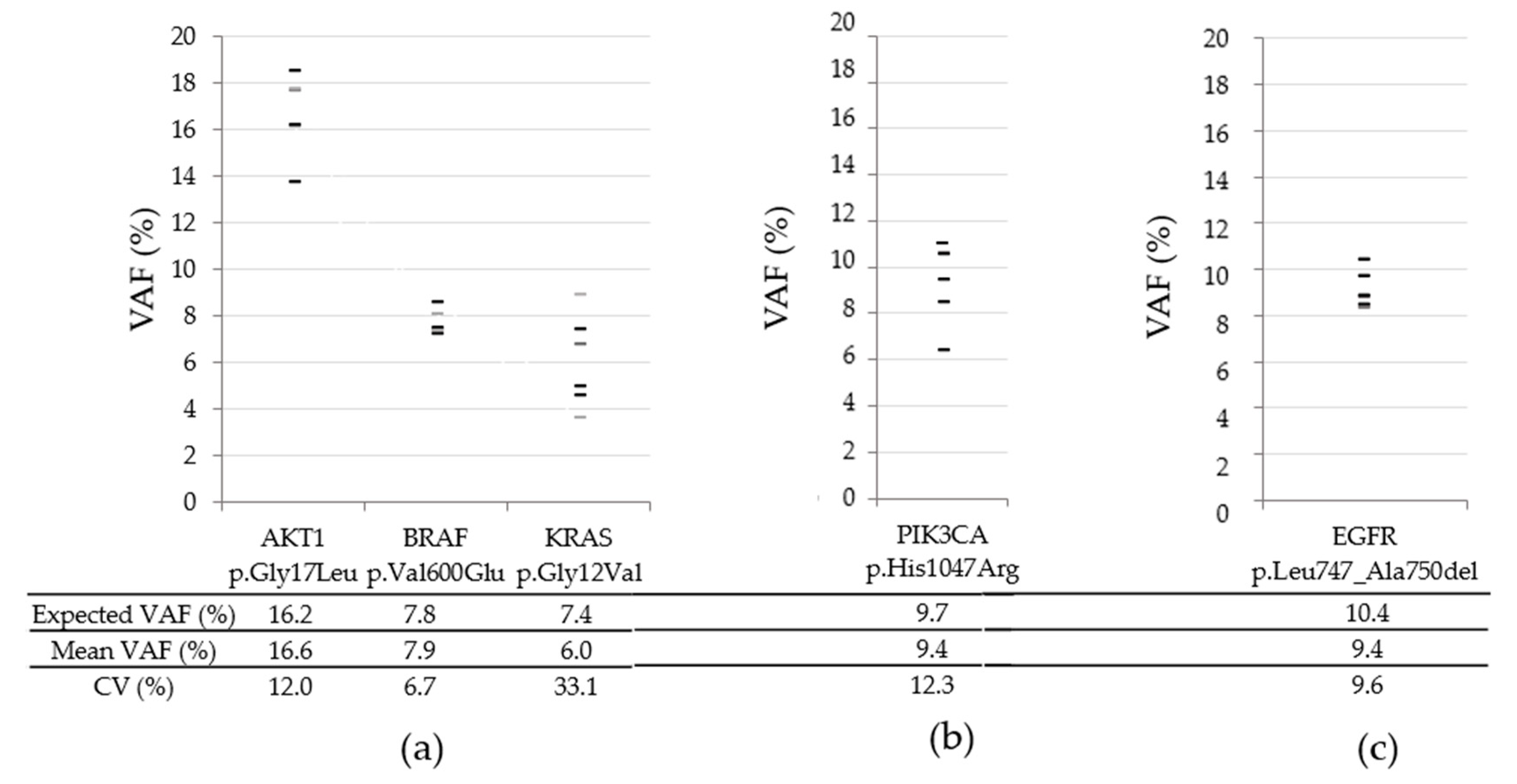
3.2. Long Term Inter-Run Variability for SNVs and Indels
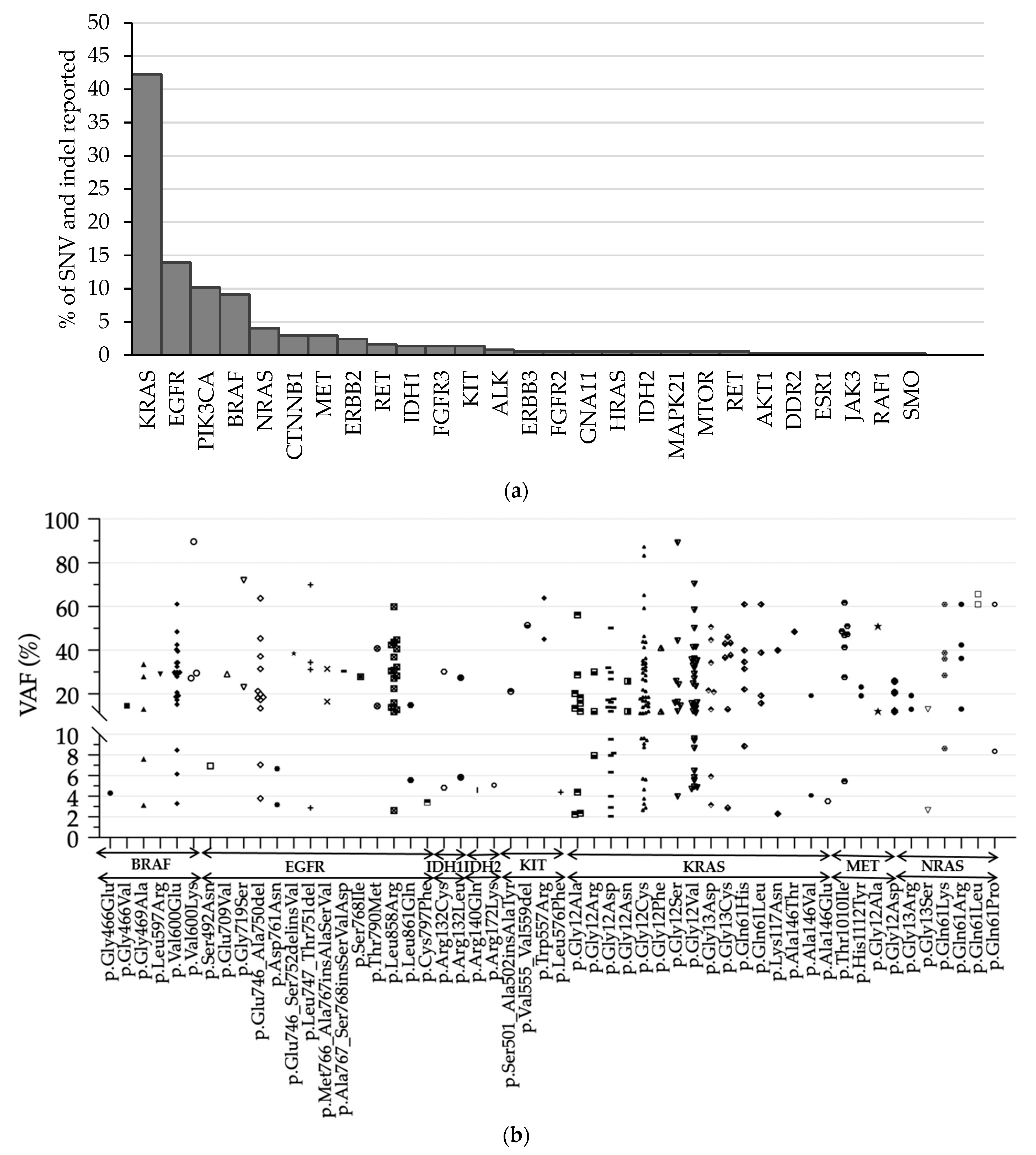
3.3. Detection of SNVs and Indels
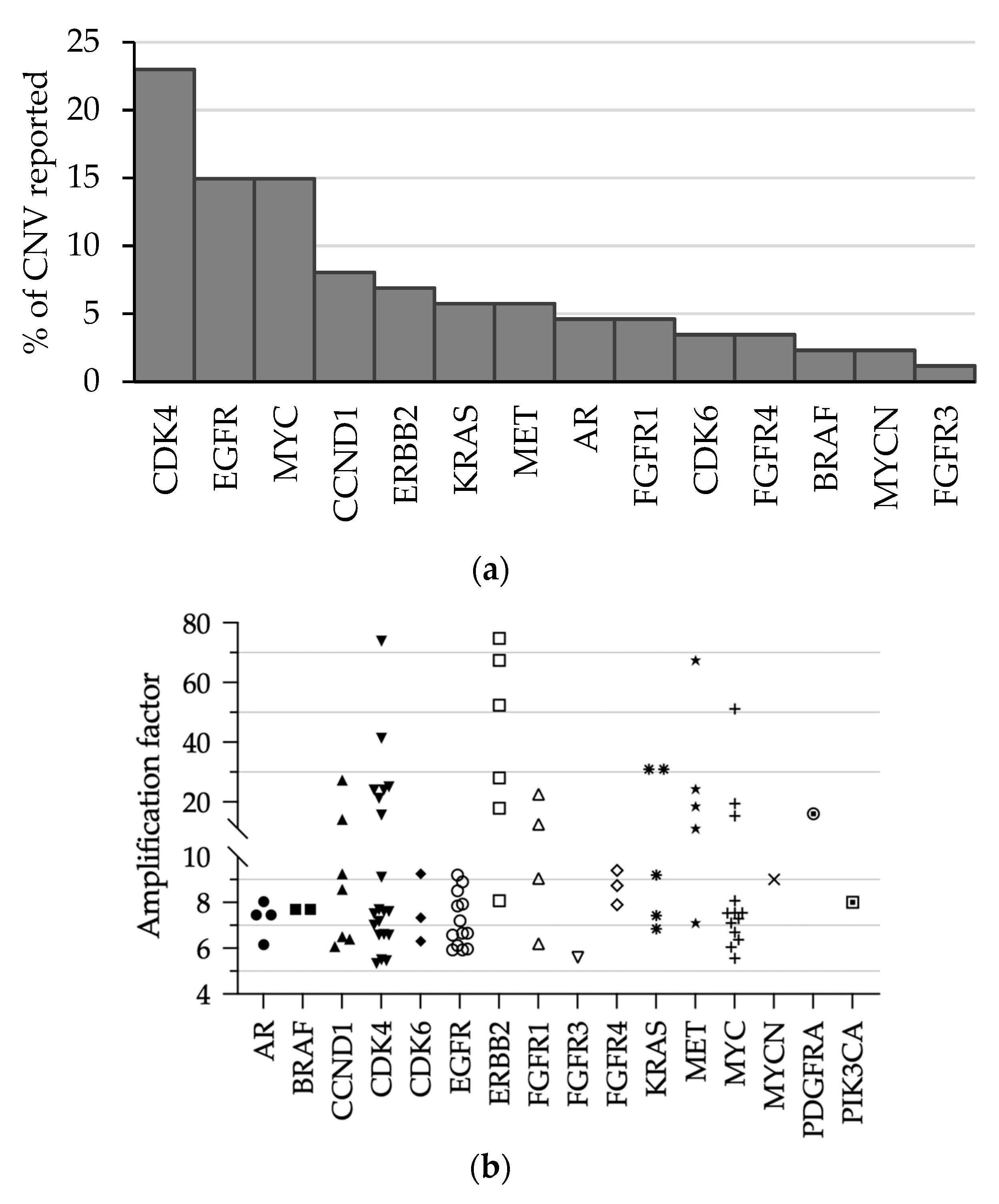
3.4. Performances of CNV Detection
3.5. Detection of Gene Amplification in Routine Practice
3.6. Gene-Fusion and Exon-Skipping Detection
3.7. Detection of Fusion and Exon Skipping in Routine Practice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Genomic position of the added clinically relevant hotspot mutations.
| Chromosome | Genomic position | Allele | Amplicon | |
| chr1 | 115252288 | 115252289 | REF = C;OBS = T | OCP1_NRAS_1 |
| chr1 | 115252288 | 115252289 | REF = C;OBS = G | OCP1_NRAS_1 |
| chr1 | 115252289 | 115252290 | REF = T;OBS = A | OCP1_NRAS_1 |
| chr1 | 115252289 | 115252290 | REF = T;OBS = C | OCP1_NRAS_1 |
| chr1 | 115252289 | 115252290 | REF = T;OBS = G | OCP1_NRAS_1 |
| chr1 | 115252290 | 115252291 | REF = T;OBS = A | OCP1_NRAS_1 |
| chr1 | 115252290 | 115252291 | REF = T;OBS = C | OCP1_NRAS_1 |
| chr1 | 115252290 | 115252291 | REF = T;OBS = G | OCP1_NRAS_1 |
| chr1 | 115256536 | 115256537 | REF = T;OBS = A | CHP2_NRAS_2 |
| chr1 | 115256536 | 115256537 | REF = T;OBS = G | CHP2_NRAS_2 |
| chr1 | 115256536 | 115256537 | REF = T;OBS = C | CHP2_NRAS_2 |
| chr1 | 115256537 | 115256538 | REF = G;OBS = C | CHP2_NRAS_2 |
| chr1 | 115256537 | 115256538 | REF = G;OBS = T | CHP2_NRAS_2 |
| chr1 | 115256537 | 115256538 | REF = G;OBS = A | CHP2_NRAS_2 |
| chr1 | 115256538 | 115256539 | REF = T;OBS = C | CHP2_NRAS_2 |
| chr1 | 115256538 | 115256539 | REF = T;OBS = A | CHP2_NRAS_2 |
| chr1 | 115256538 | 115256539 | REF = T;OBS = G | CHP2_NRAS_2 |
| chr7 | 140453137 | 140453138 | REF = T;OBS = A | CHP2_BRAF_2 |
| chr7 | 140453137 | 140453138 | REF = T;OBS = C | CHP2_BRAF_2 |
| chr7 | 140453137 | 140453138 | REF = T;OBS = G | CHP2_BRAF_2 |
| chr7 | 140453138 | 140453139 | REF = G;OBS = A | CHP2_BRAF_2 |
| chr7 | 140453138 | 140453139 | REF = G;OBS = C | CHP2_BRAF_2 |
| chr7 | 140453138 | 140453139 | REF = G;OBS = T | CHP2_BRAF_2 |
| chr7 | 140453139 | 140453140 | REF = T;OBS = A | CHP2_BRAF_2 |
| chr7 | 140453139 | 140453140 | REF = T;OBS = C | CHP2_BRAF_2 |
| chr7 | 140453139 | 140453140 | REF = T;OBS = G | CHP2_BRAF_2 |
| chr7 | 140453140 | 140453141 | REF = A;OBS = G | CHP2_BRAF_2 |
| chr7 | 140453140 | 140453141 | REF = A;OBS = T | CHP2_BRAF_2 |
| chr7 | 140453140 | 140453141 | REF = A;OBS = C | CHP2_BRAF_2 |
| chr7 | 140453140 | 140453141 | REF = A;OBS = C | CHP2_BRAF_2 |
| chr7 | 140481415 | 140481416 | REF = T;OBS = A | OCP1_BRAF_1 |
| chr7 | 140481415 | 140481416 | REF = T;OBS = G | OCP1_BRAF_1 |
| chr7 | 140481415 | 140481416 | REF = T;OBS = C | OCP1_BRAF_1 |
| chr7 | 140481416 | 140481417 | REF = C;OBS = T | OCP1_BRAF_1 |
| chr7 | 140481416 | 140481417 | REF = C;OBS = A | OCP1_BRAF_1 |
| chr7 | 140481416 | 140481417 | REF = C;OBS = G | OCP1_BRAF_1 |
| chr7 | 140481417 | 140481418 | REF = C;OBS = G | OCP1_BRAF_1 |
| chr7 | 140481417 | 140481418 | REF = C;OBS = A | OCP1_BRAF_1 |
| chr7 | 140481417 | 140481418 | REF = C;OBS = T | OCP1_BRAF_1 |
| chr12 | 25378556 | 25378557 | REF = C;OBS = A | CHP2_KRAS_3 |
| chr12 | 25378556 | 25378557 | REF = C;OBS = T | CHP2_KRAS_3 |
| chr12 | 25378556 | 25378557 | REF = C;OBS = G | CHP2_KRAS_3 |
| chr12 | 25378557 | 25378558 | REF = T;OBS = A | CHP2_KRAS_3 |
| chr12 | 25378557 | 25378558 | REF = T;OBS = C | CHP2_KRAS_3 |
| chr12 | 25378557 | 25378558 | REF = T;OBS = G | CHP2_KRAS_3 |
| chr12 | 25378558 | 25378559 | REF = T;OBS = A | CHP2_KRAS_3 |
| chr12 | 25378558 | 25378559 | REF = T;OBS = C | CHP2_KRAS_3 |
Appendix B
- Amplicon ID list:
- 1.OCP1_AR_8, 2.OCP1_DCUN1D1_5, 3.OCP1_AR_10, 4.OCP1_AR_2, 5.OCP1_DCUN1D1_9, 6.OCP1_MYCN_2, 7.Oncomine_Focus_PIK3CA_1, 8.OCP1_KRAS_10, 9.OCP1_MYCN_14, 10.OCP1_NF1_49, 11.OCP1_FGFR3_12, 12.OCP1_AR_16, 13.Oncomine_Focus_BRAF_5, 14.OCP1_KRAS_1, 15.OCP1_FGFR2_12, 16.OCP1_FGFR2_16, 17.OCP1_EGFR_13, 18.OCP1_FGFR3_3, 19.OCP1_PDGFRA_4, 20.OCP1_MED12_3, 21.OCP1_FGFR3_1, 22.OCP1_AR_6, 23.OCP1_FGFR2_8, 24.ON_DDR2_2, 25.OCP1_CDK4_2, 26.OCP1_KIT_10, 27.OCP1_EGFR_16, 28.Oncomine_Focus_BRAF_7, 29.Oncomine_Focus_ALK_1, 30.OCP1_DCUN1D1_3, 31.OCP1_AR_3, 32.OCP1_KIT_1, 33.OCP1_BRCA1_91, 34.OCP1_NF1_1, 35.OCP1_AR_12, 36.OCP1_FGFR4_5, 37.OCP1_BIRC2_11, 38.OCP1_BRCA1_84, 39.OCP1_MYC_5, 40.OCP1_MED12_1, 41.OCP1_MYCN_12, 42.OCP1_KIT_7, 43.OCP1_CDK6_2, 44.OCP1_CCND1_1, 45.OCP1_AR_11, 46.CHP2_CTNNB1_1, 47.OCP1_MYCN_19, 48.OCP1_AR_14, 49.OCP1_MYCN_10, 50.CHP2_FGFR3_3, 51.OCP1_NF1_89, 52.OCP1_PDGFRA_18, 53.CHP2_SMO_3, 54.OCP1_KRAS_4, 55.OCP1_MTOR_2, 56.OCP1_BRCA1_6, 57.OCP1_PDGFRA_10, 58.CHP2_NRAS_1, 59.OCP1_CDK6_13, 60.OCP1_MYC_3, 61.OCP1_CCND1_10, 62.OCP1_MET_13, 63.OCP1_GNAQ_1, 64.OCP1_MYC_11, 65.Oncomine_Focus_PIK3CA_2, 66.OCP1_CDK4_8, 67.OCP1_FGFR4_16, 68.Oncomine_Focus_ALK_6, 69.CHP2_PIK3CA_10, 70.OCP1_ESR1_3, 71.OCP1_AR_15, 72.OCP1_MYCN_1, 73.CHP2_FGFR2_1, 74.CHP2_FGFR3_4, 75.OCP1_ERBB2_22, 76.OCP1_MET_16, 77.ON_EGFR_2A, 78.OCP1_FGFR2_18, 79.OCP1_MET_1, 80.Oncomine_Focus_ALK_5, 81.OCP1_CCND1_6, 82.OCP1_FGFR4_17, 83.OCP1_MYCN_9, 84.OCP1_CCND1_4, 85.OCP1_CCND1_18, 86.OCP1_MTOR_4, 87.OCP1_CCND1_7, 88.OCP1_APC_32, 89.OCP1_MYC_6, 90.CCP_EGFR_6, 91.OCP1_FGFR1_2, 92.CHP2_FGFR3_2, 93.OCP1_ERBB2_14, 94.OCP1_FGFR4_13, 95.OCP1_JAK3_2, 96.OCP1_CDK4_14, 97.OCP1_PDGFRA_7, 98.OCP1_DCUN1D1_10, 99.OCP1_BIRC2_12, 100.Oncomine_Focus_MTOR_3, 101.OCP1_MET_19, 102.OCP1_PIK3CA_18, 103.Oncomine_Focus_ALK_7, 104.OCP1_APC_40, 105.CHP2_MET_5, 106.Oncomine_Focus_AR_1, 107.OCP1_KRAS_8, 108.OCP1_FGFR1_17, 109.OCP1_FGFR4_2, 110.OCP1_FGFR4_18, 111.OCP1_ALK_1, 112.OCP1_RAF1_1, 113.CHP2_GNA11_1, 114.OCP1_KRAS_5, 115.OCP1_CDK6_14, 116.OCP1_MYCN_6, 117.OCP1_FGFR1_10, 118.Oncomine_Focus_HRAS_1, 119.CHP2_KIT_5, 120.OCP1_MYC_4, 121.OCP1_BIRC2_8, 122.OCP1_FGFR3_8, 123.CHP2_SMO_4, 124.OCP1_FGFR1_18, 125.OCP1_FGFR2_13, 126.OCP1_CDK6_5, 127.OCP1_NRAS_1, 128.OCP1_BIRC2_14, 129.CHP2_RET_2, 130.ON_MAP2K1_1, 131.CHP2_IDH2_1, 132.OCP1_MET_5, 133.CHP2_PDGFRA_2, 134.CHP2_RET_3, 135.OCP1_JAK1_1, 136.OCP1_ERBB2_23, 137.CHP2_PIK3CA_6, 138.OCP1_KRAS_7, 139.Oncomine_Focus_ALK_4, 140.OCP1_CCND1_17, 141.OCP1_FGFR1_8, 142.CHP2_KRAS_3, 143.CHP2_GNAQ_1, 144.Oncomine_Focus_PIK3CA_3, 145.OCP1_FGFR1_13, 146.CHP2_BRAF_2, 147.CHP2_NRAS_2, 148.Oncomine_Focus_GNA11_1, 149.OCP1_CDK6_15, 150.CHP2_FGFR2_4, 151.OCP1_CCND1_8, 152.Oncomine_Focus_BRAF_4, 153.OCP1_MYC_1, 154.OCP1_ERBB3_1, 155.OCP1_NF1_130, 156.OCP1_MYC_2, 157.Oncomine_Focus_MTOR_2, 158.Oncomine_Focus_MET_1, 159.CHP2_FGFR1_2, 160.OCP1_ALK_2, 161.CHP2_RET_4, 162.OCP1_CDK4_6, 163.OCP1_BRCA1_88, 164.OCP1_ERBB2_12, 165.Oncomine_Focus_SMO_1, 166.Oncomine_Focus_MTOR_4, 167.OCP1_MTOR_3, 168.OCP1_APC_1, 169.CHP2_KIT_2, 170.OCP1_MAP2K1_2, 171.OCP1_CDK4_7, 172.OCP1_FGFR3_13, 173.CHP2_AKT1_1, 174.CHP2_HRAS_2, 175.Oncomine_Focus_SMO_2, 176.CHP2_RET_1, 177.Oncomine_Focus_ROS1_1, 178.OCP1_KRAS_6, 179.OCP1_KIT_13, 180.Oncomine_Focus_FGFR2_1, 181.Oncomine_Focus_FGFR2_2, 182.OCP1_CDK6_4, 183.OCP1_BRAF_1, 184.OCP1_CCND1_9, 185.CHP2_FGFR3_1, 186.OCP1_APC_62, 187.OCP1_MYCN_13, 188.OCP1_FGFR4_11, 189.CHP2_ERBB2_2, 190.OCP1_BRCA1_11, 191.OCP1_CDK4_4, 192.CHP2_MET_3, 193.OCP1_CDK6_6, 194.Oncomine_Focus_KIT_1, 195.OCP1_BIRC2_7, 196.OCP1_KIT_20, 197.Oncomine_Focus_RAF1_1, 198.OCP1_CDK4_13, 199.OCP1_MYC_7, 200.CHP2_EGFR_3, 201.OCP1_JAK3_1, 202.CHP2_EGFR_4, 203.OCP1_FGFR4_10, 204.Oncomine_Focus_MTOR_1, 205.CHP2_RET_5, 206.Oncomine_Focus_ERBB2_2, 207.OCP1_CCND1_2, 208.Oncomine_Focus_BRAF_8, 209.OCP1_DCUN1D1_12, 210.OCP1_FGFR4_8, 211.Oncomine_Focus_ROS1_2, 212.OCP1_CDK4_12, 213.Oncomine_Focus_BRAF_6, 214.CHP2_PIK3CA_9, 215.CHP2_MET_6, 216.OCP1_MAP2K2_1, 217.OCP1_MYC_10, 218.OCP1_FGFR3_5, 219.Oncomine_Focus_BRAF_3, 220.CHP2_EGFR_2, 221.OCP1_ERBB2_17, 222.Oncomine_Focus_BRAF_1, 223.OCP1_FGFR1_7, 224.ON_ALK_0, 225.CHP2_KIT_3, 226.CHP2_FGFR2_3, 227.Oncomine_Focus_ERBB3_1, 228.CHP2_PDGFRA_1, 229.CHP2_JAK2_1, 230.OCP1_CDK4_3, 231.Oncomine_Focus_ALK_2, 232. Oncomine_Focus_JAK1_1, 233. OCP1_JAK1_2, 234. OCP1_FGFR1_16, 235. Oncomine_Focus_ERBB2_1, 236. OCP1_PDGFRA_6, 237. OCP1_MYC_8, 238. OCP1_MET_8, 239. OCP1_FGFR4_4, 240. OCP1_PIK3CA_19, 241. CHP2_EGFR_1, 242. OCP1_ERBB3_2, 243. OCP1_CDK6_12, 244. CHP2_PIK3CA_3, 245. OCP1_PDGFRA_12, 246. OCP1_FGFR1_1, 247. CHP2_KRAS_1, 248. OCP1_CDK6_7, 249. Oncomine_Focus_JAK3_1, 250. Oncomine_Focus_ALK_3, 251. OCP1_PDGFRA_2, 252. OCP1_EGFR_23, 253. Oncomine_Focus_BRAF_2, 254. CHP2_IDH1_1, 255. OCP1_MAP2K1_1, 256. OCP1_MYCN_8, 257. Oncomine_Focus_ERBB4_1, 258. OCP1_PIK3CA_20, 259. CHP2_ERBB2_3, 260. OCP1_CDK6_11, 261. Oncomine_Focus_ERBB3_2, 262. OCP1_APC_21, 263. Oncomine_Focus_ERBB3_3, 264. CHP2_PDGFRA_4, 265. OCP1_CDK4_10, 266. CHP2_KRAS_2, 267. CHP2_ERBB2_1, 268. CHP2_EGFR_5, 269. CHP2_KIT_4.
References
- Caputo, V.; De Falco, V.; Ventriglia, A.; Famiglietti, V.; Martinelli, E.; Morgillo, F.; Martini, G.; Corte, C.M.D.; Ciardiello, D.; Poliero, L.; et al. Comprehensive genome profiling by next generation sequencing of circulating tumor DNA in solid tumors: A single academic institution experience. Ther. Adv. Med. Oncol. 2022, 14, 175883592210968. [Google Scholar] [CrossRef] [PubMed]
- Chougule, A.; Jagtap, V.; Nikam, A.; Kale, S.; Nambiar, K.; Bagayatkar, P.; Chandrani, P.; Kaushal, R.; Noronha, V.; Patil, V.; et al. Comprehensive Development and Implementation of Good Laboratory Practice for NGS Based Targeted Panel on Solid Tumor FFPE Tissues in Diagnostics. Diagnostics 2022, 12, 1291. [Google Scholar] [CrossRef] [PubMed]
- So, M.-K.; Park, J.-H.; Kim, J.-W.; Jang, J.-H. Analytical Validation of a Pan-Cancer Panel for Cell-Free Assay for the Detection of EGFR Mutations. Diagnostics 2021, 11, 1022. [Google Scholar] [CrossRef] [PubMed]
- 14:00–17:00 ISO 15189:2012. Available online: https://www.iso.org/cms/render/live/fr/sites/isoorg/contents/data/standard/05/61/56115.html (accessed on 19 October 2022).
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels. J. Mol. Diagn. JMD 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, 59. Available online: https://onlinelibrary.wiley.com/doi/10.1002/cpmb (accessed on 15 February 2023). [CrossRef] [PubMed]
- Ottestad, A.L.; Huang, M.; Emdal, E.F.; Mjelle, R.; Skarpeteig, V.; Dai, H.Y. Assessment of Two Commercial Comprehensive Gene Panels for Personalized Cancer Treatment. J. Pers. Med. 2022, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.L.; Walsh, K.; Diamond, A.; Oniscu, A.; Deans, Z.C. Validation of the OncomineTM focus panel for next-generation sequencing of clinical tumour samples. Virchows Arch. Int. J. Pathol. 2018, 473, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, J.; Amemiya, Y.; Arts, H.; Bayani, J.; Eng, B.; Grafodatskaya, D.; Kamel Reid, S.; Lariviere, M.; Lo, B.; McClure, R.; et al. Multisite verification of the accuracy of a multi-gene next generation sequencing panel for detection of mutations and copy number alterations in solid tumours. PLoS ONE 2021, 16, e0258188. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M.; Sun, H.; Butaney, M.; Zhang, C.; Lee, C.; Jänne, P.A.; Rodig, S.J. ROS1 Immunohistochemistry for Detection of ROS1-Rearranged Lung Adenocarcinomas. Am. J. Surg. Pathol. 2013, 37. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chung, J.-H. Overview of clinicopathologic features of ALK-rearranged lung adenocarcinoma and current diagnostic testing for ALK rearrangement. Transl. Lung Cancer Res. 2015, 4, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Dong, F.; Borger, D.; Dias-Santagata, D.; Ellisen, L.W.; Le, L.P.; Iafrate, A.J. Standardized decision support in next generation sequencing reports of somatic cancer variants. Mol. Oncol. 2014, 8, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, R.; Li, Z.; Ding, J.; Xie, J.; Li, J. Challenges of Providing Concordant Interpretation of Somatic Variants in Non-Small Cell Lung Cancer: A Multicenter Study. J. Cancer 2019, 10, 1814–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanno, L.; Pavan, A.; Ferro, A.; Calvetti, L.; Frega, S.; Pasello, G.; Aprile, G.; Guarneri, V.; Conte, P.; on behalf of Rete Oncologica Veneta (ROV). Clinical Impact of Plasma and Tissue Next-Generation Sequencing in Advanced Non-Small Cell Lung Cancer: A Real-World Experience. Oncologist 2020, 25, e1996–e2005. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Tran, H.; Eng, K.W.; Ramazanoglu, S.; Marrero Rolon, R.M.; Scognamiglio, T.; Borczuk, A.; Mosquera, J.M.; Pan, Q.; Sboner, A.; et al. Performance Characteristics of a Targeted Sequencing Platform for Simultaneous Detection of Single Nucleotide Variants, Insertions/Deletions, Copy Number Alterations, and Gene Fusions in Cancer Genome. Arch. Pathol. Lab. Med. 2020, 144, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro-Sibilot, D.; Cozic, N.; Pérol, M.; Mazières, J.; Otto, J.; Souquet, P.J.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.D.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: Results of the AcSé phase II trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1985–1991. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oncomine Focus Assay | Tumor Hot Spot | ||||||
|---|---|---|---|---|---|---|---|
| Expected VAF (%) | n | Mean VAF (%) (Mean ± SD) | CV (%) | N | Mean VAF (%) (Mean ± SD) | CV (%) | |
| BRAF p.Val600Glu | 11 | 42 | 11.8 (11.1–12.5) | 6.0 | 43 | 11.6 (9.8–13.4) | 7.7 |
| EGFR p.Glu746_Ala750del | 2 | 42 | 1.5 (1.2–1.9) | 22.4 | 43 | 1.9 (1.6–2.3) | 19.2 |
| EGFR p.Glu746_Ala750del | 5 | 35 | 4.0 (3.4–4.5) | 14.5 | 42 | 5.1 (4.5–5.7) | 11.5 |
| EGFR p.Gly719Ser | 5 | 35 | 5.2 (4.5–6.0) | 14.6 | 42 | 5.7 (5.2–6.2) | 9.4 |
| EGFR p.Thr790Met | 1 | 42 | 1.1 (0.7–1.5) | 37.7 | 43 | 1.2 (0.6–1.7) | 46.5 |
| EGFR p.Leu861Gln | 5 | 35 | 5.5 (4.9–6.0) | 10.3 | 42 | 4.7 (4.3–5.2) | 10.0 |
| EGFR p.Leu858Arg | 3 | 42 | 3.4 (2.7–3.9) | 13.3 | 43 | 3.4 (3.0–4.0) | 14.1 |
| KRAS p.Gly12Asp | 6 | 42 | 6.2 (5.5–7.0) | 12.3 | 43 | 6.5 (4.8–8.7) | 12.9 |
| KRAS p.Gly13Asp | 15 | 42 | 14.2 (13.1–15.4) | 8.1 | 43 | 15.2 (11.7–18.7) | 11.5 |
| NRAS p.Gln61Lys | 13 | 42 | 12.9 (11.6–14.2) | 9.8 | 43 | 11.8 (9.4–14.1) | 10.0 |
| PIK3CA p.Glu545Lys | 9 | 42 | 7.5 (5.4–9.5) | 11.9 | 43 | 8.3 (6.7–10.0) | 10.1 |
| PIK3CA p.His1047Arg | 18 | 42 | 17.5 (16.4–18.4) | 5.6 | 43 | 17.7 (15.3–20.1) | 6.8 |
| Gene | Expected CNV | Measured CNV (Mean ± SD) | CV (%) | 5% Limit Variability of OFA Confidence Interval (Mean ± SD) | 95% Limit Variability of OFA Confidence Interval (Mean ± SD) |
|---|---|---|---|---|---|
| MET | 4.5 | 5.8 ± 0.3 | 5.2% | 4.5 ± 0.4 | 7.4 ± 0.4 |
| MYCN | 8.5 | 14.1 ± 1.0 | 3.7% | 11.3 ± 1.0 | 17.6 ± 0.8 |
| Sample | MAPD | ERBB2 CNV | CNV Confidence Interval (5–95%) | Immunochemistry Staining Intensity |
|---|---|---|---|---|
| Patient 1 | 0.323 | 28.0 | 22.2–35.2 | Strong (+++) |
| Patient 2 | 0.360 | 9.5 | 7.5–12.0 | Strong (+++) |
| Patient 3 | 0.282 | 5.4 | 4.2–6.8 | Intermediate (++/+++) |
| Patient 4 | 0.334 | 12.0 | 8.3–16.5 | Low (+) |
| HD784 QC Material (n =29) | ||||
| Total Mapped Fusion Allele Reads: 95,753 (57,060–134,445); CV: 40.4% | ||||
| Fusion Reads Counts | Fusion Reads (%) | |||
| Mean (Mean ± SD) | CV (%) | Mean (Mean ± SD) | CV (%) | |
| CCDC6(1)-RET(12) | 4034 (1867–6212) | 54.0 | 4.6 (3.5–5.7) | 23.2 |
| SLC34A2(4)-ROS1(32) | 14,637 (7927–21,348) | 45.8 | 17.8 (13.0–22.7) | 27.1 |
| EML4(13)-ALK(20) | 4289 (1457–7121) | 35.2 | 4.5 (2.9–6.1) | 27.2 |
| Seracare QC Material (n =7) | ||||
| Total Mapped Fusion Allele Reads: 98,353 (73,474–123,233); CV: 23.3% | ||||
| Fusion Reads Counts | Fusion Reads (%) | |||
| Mean (Mean ± SD) | CV (%) | Mean (Mean ± SD) | CV (%) | |
| FGFR3(17)-BAIAP2L1(2) | 4436 (3849–5023) | 13.2 | 4.6 (3.9–5.3) | 15.1 |
| CCDC6(1)-RET(12) | 1589 (1194–1985) | 24.9 | 1.6 (1.4–1.9) | 17.5 |
| KIF5B(24)-RET(11) | 3639 (24,511–4828) | 32.7 | 3.7 (2.9–4.5) | 21.8 |
| NCOA4(8)-RET(12) | 1706 (1458–1955) | 14.6 | 1.8 (1.6–1.9) | 9.5 |
| TMPRSS2(1)-ERG(2) | 3282 (1437–5126) | 56.2 | 3.2 (2.2–4.2) | 31.1 |
| PAX8(9)-PPARG(3) | 3909 (2476–5343) | 36.7 | 3.9 (3.2–4.7) | 18.1 |
| EML4(13)-ALK(20) | 2837 (2464–3211) | 13.2 | 3 (2.3–3.7) | 22.2 |
| SLC45A3(1)-BRAF(8) | 5436 (1681–9192) | 69.1 | 5.2 (3.3–7.0) | 36.0 |
| SLC34A2(4) -ROS1(34) | 3437 (2111–4761) | 38.6 | 3.4 (2.9–4.0) | 15.9 |
| CD74(6)-ROS1(34) | 7090 (5700–8479) | 19.6 | 7.4 (5.8–8.9) | 20.9 |
| LMNA(2)-NTRK1(10) | 1701 (531–2874) | 68.8 | 1.6 (1.0–2.6) | 38.4 |
| TFG(5)-NTRK1(10) | 1323 (923–1723) | 30.2 | 1.3 (1.4–1.5) | 14.9 |
| TPM3(7)-NTRK1(9) | 2822 (2127–3517) | 24.6 | 2.9 (2.6–3.1) | 7.9 |
| ETV6(5)-NTRK3(15) | 2352 (1908–2796) | 18.9 | 2.4 (2.0–2.9) | 17.4 |
| EGFR(1)-EGFR(8) (vIII) | 2605 (1390–3821) | 46.6 | 2.6 (2.0–3.0) | 18.3 |
| MET(13)-MET(15) | 2451 (2012–2889) | 17.9 | 2.6 (1.9–3.3) | 27.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamba-Funck, J.; Fabre, E.E.; Kambouchner, M.; Schischmanoff, O. Performance Characteristics of Oncomine Focus Assay for Theranostic Analysis of Solid Tumors, A (21-Months) Real-Life Study. Diagnostics 2023, 13, 937. https://doi.org/10.3390/diagnostics13050937
Bamba-Funck J, Fabre EE, Kambouchner M, Schischmanoff O. Performance Characteristics of Oncomine Focus Assay for Theranostic Analysis of Solid Tumors, A (21-Months) Real-Life Study. Diagnostics. 2023; 13(5):937. https://doi.org/10.3390/diagnostics13050937
Chicago/Turabian StyleBamba-Funck, Jessica, Emmanuelle E. Fabre, Marianne Kambouchner, and Olivier Schischmanoff. 2023. "Performance Characteristics of Oncomine Focus Assay for Theranostic Analysis of Solid Tumors, A (21-Months) Real-Life Study" Diagnostics 13, no. 5: 937. https://doi.org/10.3390/diagnostics13050937
APA StyleBamba-Funck, J., Fabre, E. E., Kambouchner, M., & Schischmanoff, O. (2023). Performance Characteristics of Oncomine Focus Assay for Theranostic Analysis of Solid Tumors, A (21-Months) Real-Life Study. Diagnostics, 13(5), 937. https://doi.org/10.3390/diagnostics13050937