
Proteomic Approaches and Potential Applications in Autosomal Dominant Polycystic Kidney Disease and Fabry Disease

Abstract
:1. Introduction
Proteomics
2. ADPKD
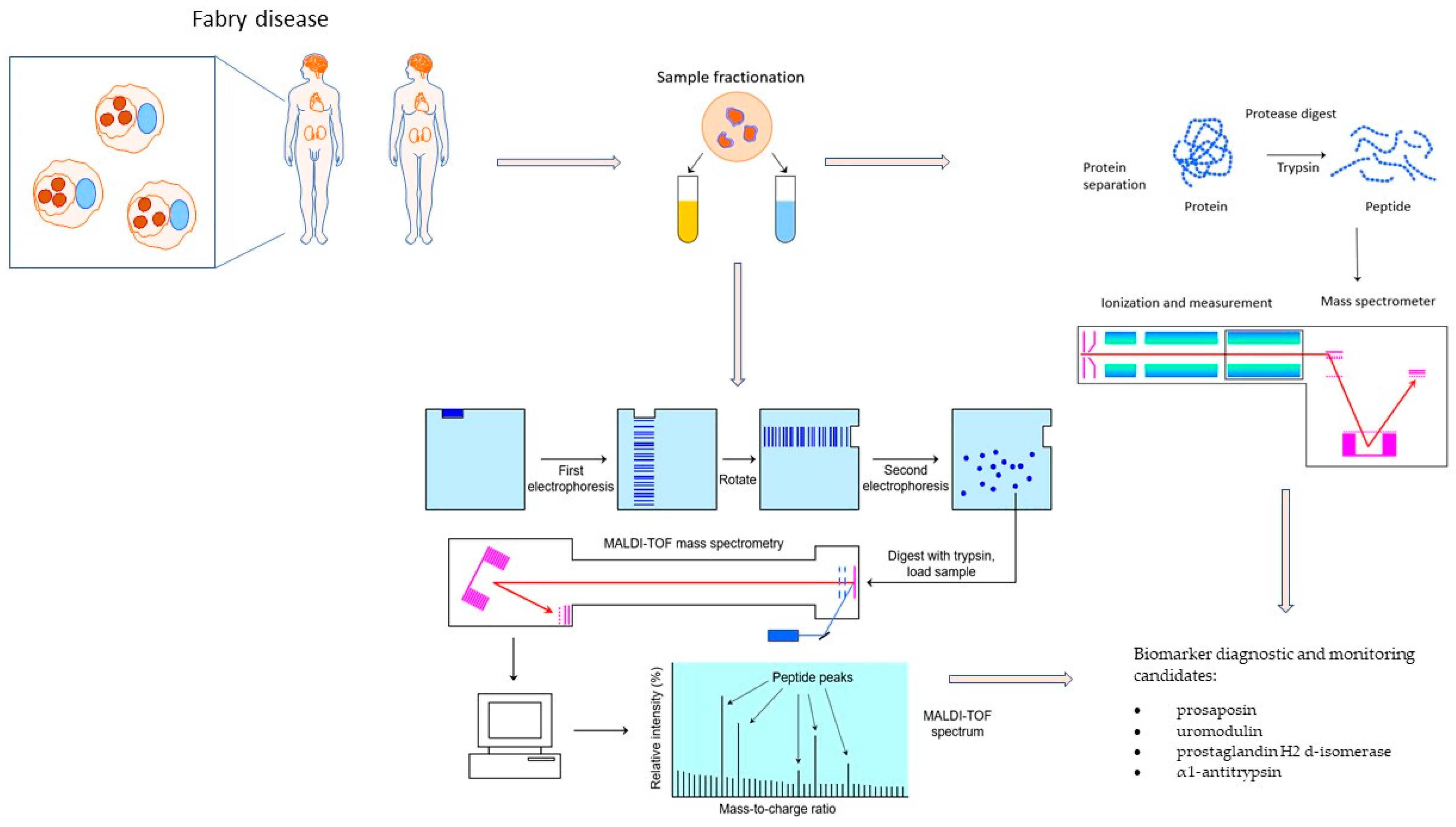
3. Fabry Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- Torra, R.; Furlano, M.; Ortiz, A.; Ars, E. Genetic kidney diseases as an underrecognized cause of chronic kidney disease: The key role of international registry reports. Clin. Kidney J. 2021, 14, 1879–1885. [Google Scholar] [CrossRef]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef]
- Groopman, E.E.; Rasouly, H.M.; Gharavi, A.G. Kidney genomics. Nat. Rev. Nephrol. 2018, 14, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Köttgen, A.; Cornec-Le Gall, E.; Halbritter, J.; Kiryluk, K.; Mallett, J.A.; Parekh, S.R.; Rasouly, M.H.; Sampson, G.M.; Tin, A.; Antignac, C.; et al. Genetics in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2022, 101, 1126–1141. [Google Scholar] [CrossRef]
- Rodríguez, R.M.; Luis-Lima, S.; Fernandez, J.M.; Gómez, M.V.P.; Toledo, B.G.; Cobo, M.; Delgado-Mallén, P.; Escamilla, B.; Marco, C.O.; Estupiñán, S.; et al. Estimated GFR in autosomal dominant polycystic kidney disease: Errors of an unpredictable method. J. Nephrol. 2022, 35, 2109–2118. [Google Scholar] [CrossRef]
- Raby, K.L.; Horsely, H.; McCarthy-Boxer, A.; Norman, J.T.; Wilson, P.D. Urinary exosome proteomic profiling defines stage-specific rapid progression of autosomal dominant polycystic kidney disease and tolvaptan efficacy. BBA Adv. 2021, 1, 100013. [Google Scholar] [CrossRef]
- Bruschi, M.; Granata, S.; Candiano, G.; Fabris, A.; Petretto, A.; Ghiggeri, G.M.; Gambaro, G.; Zaza, G. Proteomic analysis of urinary extracellular vesicles reveals a role for the complement system in medullary sponge kidney disease. Int. J. Mol. Sci. 2019, 20, 5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirolli, V.; Pieroni, L.; Di Liberato, L.; Urbani, A.; Bonomini, M. Urinary peptidomic biomarkers in kidney diseases. Int. J. Mol. Sci. 2020, 21, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantari, S.; Jafari, A.; Moradpoor, R.; Ghasemi, E.; Khalkhal, E. Human Urine Proteomics: Analytical Techniques and Clinical Applications in Renal Diseases. Int. J. Proteom. 2015, 2015, 782798. [Google Scholar] [CrossRef]
- Merchant, M.L. Mass Spectrometry in Chronic Kidney Disease Research. Adv. Chronic Kidney Dis. 2010, 17, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Pejchinovski, M.; Magalhães, P.; Metzeger, J. Editorial: Clinical application of proteomics in kidney diseases. Front. Med. 2022, 9, 965083. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, K.J.R.; Dittrich, S.; Bartram, M.P.; Rinschen, M.M. Quantification of molecular heterogeneity in kidney tissue by targeted proteomics. J. Proteom. 2019, 193, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Dubin, R.F.; Rhee, E.P. Proteomics and metabolomics in kidney disease, including insights into etiology, treatment, and prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Pejchinovski, M.; Mischak, H. Clinical Proteomics in Kidney Disease: From Discovery to Clinical Application. Prilozi 2017, 38, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Rinschen, M.M.; Saez-Rodriguez, J. The tissue proteome in the multi-omic landscape of kidney disease. Nat. Rev. Nephrol. 2021, 17, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Hao, L.; Ricke, W.A.; Li, L. Proteomics Clinical Apps—2015—Thomas—Biomarker discovery in mass spectrometry-based urinary proteomics. Proteom. –Clin. Appl. 2016, 10, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dear, J.W.; Street, J.M.; Bailey, M.A. Urinary exosomes: A reservoir for biomarker discovery and potential mediators of intrarenal signalling. Proteomics 2013, 13, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Salih, M.; Zietse, R.; Hoorn, E.J. Urinary extracellular vesicles and the kidney: Biomarkers and beyond. Am. J. Physiol. -Ren. Physiol. 2014, 306, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. BioScience 2015, 65, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Sun, I.O.; Lerman, L.O. Urinary extracellular vesicles as biomarkers of kidney disease: From diagnostics to therapeutics. Diagnostics 2020, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiterová, J.; Tesař, V. Autosomal Dominant Polycystic Kidney Disease: From Pathophysiology of Cystogenesis to Advances in the Treatment. Int. J. Mol. Sci. 2022, 23, 3317. [Google Scholar] [CrossRef]
- Nobakht, N.; Hanna, R.M.; Al-Baghdadi, M.; Ameen, K.M.; Arman, F.; Nobahkt, E.; Kamgar, M.; Rastogi, A. Advances in Autosomal Dominant Polycystic Kidney Disease: A Clinical Review. Kidney Med. 2020, 2, 196–208. [Google Scholar] [CrossRef]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, eaat9819. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, V.Y.; Sultanova, R.F.; Sudarikova, A.V.; Ilatovskaya, D.V. Insights into the Molecular Mechanisms of Polycystic Kidney Diseases. Front. Physiol. 2021, 12, 693130. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Müller, R.U.; Benzing, T. Cystic kidney diseases from the adult nephrologist’s point of view. Front. Pediatr. 2018, 6, 65. [Google Scholar] [CrossRef] [Green Version]
- Tangri, N.; Hougen, I.; Alam, A.; Perrone, R.; McFarlane, P.; Pei, Y. Total kidney volume as a biomarker of disease progression in autosomal dominant polycystic kidney disease. Can. J. Kidney Health Dis. 2017, 4, 2054358117693355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EAF co-chairs; Harris, T.; Sandford, R.; EAF members; Roundtable participants. European ADPKD Forum multidisciplinary position statement on autosomal dominant polycystic kidney disease care: European ADPKD Forum and Multispecialist Roundtable participants. Nephrol. Dial. Transplant. 2018, 33, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistler, A.D.; Mischak, H.; Poster, D.; Dakna, M.; Wüthrich, R.P.; Serra, A.L. Identification of a unique urinary biomarker profile in patients with autosomal dominant polycystic kidney disease. Kidney Int. 2009, 76, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Ramasubbu, K.; Gretz, N.; Bachmann, S. Increased epithelial cell proliferation and abnormal extracellular matrix in rat polycystic kidney disease. J. Am. Soc. Nephrol. 1998, 9, 937–945. [Google Scholar] [CrossRef]
- Obermüller, N.; Morente, N.; Kränzlin, B.; Gretz, N.; Witzgall, R. A possible role for metalloproteinases in renal cyst development. Am. J. Physiol. -Ren. Physiol. 2001, 280, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Peleg, I.; Greenfeld, Z.; Cooperman, H.; Shoshan, S. Type I and Type III Collagen mRNA Levels in Kidney Regions of Old and Young Rats. Matrix 1993, 13, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.C.; Gorry, M.C.; Hart, P.S.; Woodard, A.S.; Shihabi, Z.; Sandhu, J.; Shirts, B.; Xu, L.; Zhu, H.; Barmada, M.M.; et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J. Med. Genet. 2002, 39, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistler, A.D.; Serra, A.L.; Siwy, J.; Poster, D.; Krauer, F.; Torres, V.E.; Mrug, M.; Grantham, J.J.; Bae, K.T.; Bost, J.E.; et al. Urinary Proteomic Biomarkers for Diagnosis and Risk Stratification of Autosomal Dominant Polycystic Kidney Disease: A Multicentric Study. PLoS ONE 2013, 8, e53016. [Google Scholar] [CrossRef]
- Schieren, G.; Rumberger, B.; Klein, M.; Kreutz, C.; Wilpert, J.; Geyer, M.; Faller, D.; Timmer, J.; Quack, I.; Rump, L.C.; et al. Gene profiling of polycystic kidneys. Nephrol. Dial. Transplant. 2006, 21, 1816–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowley, B.D.; Ricardo, S.D.; Nagao, S.; Diamond, J.R. Increased renal expression of monocyte chemoattractant protein-1 and osteopontin in ADPKD in rats. Kidney Int. 2001, 60, 2087–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, J.; Kirsch, T.; Schiffer, E.; Ulger, P.; Mentes, E.; Brand, K.; Weissinger, E.M.; Haubitz, M.; Mischak, H.; Herget-Rosenthal, S. Urinary excretion of twenty peptides forms an early and accurate diagnostic pattern of acute kidney injury. Kidney Int. 2010, 78, 1252–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weimbs, T. Polycystic kidney disease and renal injury repair: Common pathways, fluid flow, and the function of polycystin-1. Am. J. Physiol. -Ren. Physiol. 2007, 293, F1423–F1432. [Google Scholar] [CrossRef] [Green Version]
- Bolignano, D.; Coppolino, G.; Campo, S.; Aloisi, C.; Nicocia, G.; Frisina, N.; Buemi, M. Neutrophil gelatinase-associated lipocalin in patients with autosomal-dominant polycystic kidney disease. Am. J. Nephrol. 2007, 27, 373–378. [Google Scholar] [CrossRef]
- Kuehn, E.W.; Hirt, M.N.; John, A.K.; Muehlenhardt, P.; Boehlke, C.; Pütz, M.; Kramer-Zucker, A.G.; Bashkurov, M.; van de Weyer, P.S.; Kotsis, F.; et al. Kidney injury molecule 1 (Kim1) is a novel ciliary molecule and interactor of polycystin 2. Biochem. Biophys. Res. Commun. 2007, 364, 861–866. [Google Scholar] [CrossRef]
- Pejchinovski, M.; Siwy, J.; Metzger, J.; Dakna, M.; Mischak, H.; Klein, J.; Jankowski, V.; Bae, K.T.; Chapman, A.B.; Kistler, A.D. Urine peptidome analysis predicts risk of end-stage renal disease and reveals proteolytic pathways involved in autosomal dominant polycystic kidney disease progression. Nephrol. Dial. Transplant. 2017, 32, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Nagao, S.; Yamaguchi, T.; Kusaka, M.; Maser, R.L.; Takahashi, H.; Cowley, B.D.; Grantham, J.J. Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int. 2003, 63, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Rauniyar, N.; Yu, X.; Cantley, J.; Belcher, J.; Colangelo, C.M.; Stone, K.L.; Dahl, N.; Parikh, C.; Lam, T.T. Quantification of Urinary Protein Biomarkers of Autosomal Dominant Polycystic Kidney Disease by Parallel Reaction Monitoring. Proteom. –Clin. Appl. 2018, 12, e1700157. [Google Scholar] [CrossRef]
- Hogan, M.C.; Manganelli, L.; Woollard, J.R.; Masyuk, A.I.; Masyuk, T.V.; Tammachote, R.; Huang, B.Q.; Leontovich, A.A.; Beito, T.G.; Madden, B.J.; et al. Characterization of PKD protein-positive exosome-like vesicles. J. Am. Soc. Nephrol. 2009, 20, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Hogan, M.C.; Bakeberg, J.L.; Gainullin, V.G.; Irazabal, M.V.; Harmon, A.J.; Lieske, J.C.; Charlesworth, M.C.; Johnson, K.L.; Madden, B.J.; Zenka, R.M.; et al. Identification of biomarkers for PKD1 using urinary exosomes. J. Am. Soc. Nephrol. 2015, 26, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, J.M.; Birkhoff, W.; Menzies, R.I.; Webb, D.J.; Bailey, M.A.; Dear, J.W. Exosomal transmission of functional aquaporin 2 in kidney cortical collecting duct cells. J. Physiol. 2011, 589, 6119–6127. [Google Scholar] [CrossRef]
- Pocsfalvi, G.; Raj, D.A.A.; Fiume, I.; Vilasi, A.; Trepiccione, F.; Capasso, G. Urinary extracellular vesicles as reservoirs of altered proteins during the pathogenesis of polycystic kidney disease. Proteom. –Clin. Appl. 2015, 9, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Salih, M.; Demmers, J.A.; Bezstarosti, K.; Leonhard, W.N.; Losekoot, M.; van Kooten, C.; Gansevoort, R.T.; Peters, D.J.; Zietse, R.; Hoorn, E.J.; et al. Proteomics of urinary vesicles links plakins and complement to polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 3079–3092. [Google Scholar] [CrossRef] [Green Version]
- Tomar, A.; George, S.; Kansal, P.; Wang, Y.; Khurana, S. Interaction of Phospholipase C-γ1 with Villin Regulates Epithelial Cell Migration. J. Biol. Chem. 2006, 281, 31972–31986. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; De Pascalis, C.; Distefano, G.; Ducano, N.; Oldani, A.; Lanzetti, L.; Boletta, A. Regulation of the microtubular cytoskeleton by Polycystin-1 favors focal adhesions turnover to modulate cell adhesion and migration. BMC Cell Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jefferson, J.J.; Leung, C.L.; Liem, R.K.H. Plakins: Goliaths that link cell junctions and the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2004, 5, 542–553. [Google Scholar] [CrossRef]
- Silberberg, M.; Charron, A.J.; Bacallao, R.; Wandinger-Ness, A. Mispolarization of desmosomal proteins and altered intercellular adhesion in autosomal dominant polycystic kidney disease. Am. J. Physiol. - Ren. Physiol. 2005, 288, 1–21. [Google Scholar] [CrossRef]
- Su, Z.; Wang, X.; Gao, X.; Pan, C.; Hu, H.; Beyer, R.P.; Shi, M.; Zhou, J.; Zhang, J.; Serra, A.L.; et al. Excessive activation of the alternative complement pathway in autosomal dominant polycystic kidney disease. J. Intern. Med. 2014, 276, 470–485. [Google Scholar] [CrossRef]
- Liu, J. CREG1 Interacts with Sec8 to Promote Cardiomyogenic Differentiation and Cell-Cell Adhesion. Stem Cells 2018, 36, 130. [Google Scholar] [CrossRef] [Green Version]
- Luyten, A.; Su, X.; Gondela, S.; Chen, Y.; Rompani, S.; Takakura, A.; Zhou, J. Aberrant regulation of planar cell polarity in polycystic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1521–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germino, G.G.; Guay-Woodford, L.M. Polycystic Kidney Disease. In Chronic Renal Disease; Elsevier Inc.: Amsterdam, The Netherlands, 2014; pp. 484–500. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.H.; Kang, E.; Kim, Y.M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.H.; Kim, J.M.; Choi, I.H.; et al. Fabry disease: Characterisation of the plasma proteome pre- and post-enzyme replacement therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Bertoldi, G.; Carraro, G.; Ravarotto, V.; Di Vico, V.; Anastasio, P.B.; Vitturi, N.; Francini, F.; Stefanelli, F.L.; Calo, L.A. The effect of green tea as an adjuvant to enzyme replacement therapy on oxidative stress in Fabry disease: A pilot study. Front Nutr. 2022, 9, 924710. [Google Scholar] [CrossRef]
- Liu, H.C.; Lin, H.Y.; Yang, C.F.; Liao, H.C.; Hsu, T.R.; Lo, C.W.; Chang, F.P.; Huang, C.K.; Lu, Y.H.; Lin, S.P.; et al. Globotriaosylsphingosine (lyso-Gb3) might not be a reliable marker for monitoring the long-term therapeutic outcomes of enzyme replacement therapy for late-onset Fabry patients with the Chinese hotspot mutation (IVS4+919G>A). Orphanet J. Rare Dis. 2014, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early markers of Fabry disease revealed by proteomics. Mol. BioSyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in PBMC isolated from patients with Fabry disease: Preliminary findings. Mol. BioSyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef]
- Gieselmann, R.; Kwiatkowski, D.J.; Janmey, P.A.; Witke, W. Distinct Biochemical Characteristics of the Two Human Profilin Isoforms. Eur. J. Biochem. 1995, 229, 621–628. [Google Scholar] [CrossRef]
- Tebani, A.; Mauhin, W.; Abily-donval, L.; Lesueur, C.; Berger, M.G.; Nadjar, Y.; Berger, J.; Benveniste, O.; Lamari, F.; Laforêt, P.; et al. A Proteomics-Based Analysis Reveals Predictive Biological Patterns in Fabry Disease. J. Clin. Med. 2020, 9, 1325. [Google Scholar] [CrossRef]
- Murakami, M.; Simons, M. Fibroblast growth factor regulation of neovascularization. Curr. Opin. Hematol. 2008, 15, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Choi, E.N.; Jeon, Y.J.; Jung, S.C. Possible role of transforming growth factor-β1 and vascular endothelial growth factor in Fabry disease nephropathy. Int. J. Mol. Med. 2012, 30, 1275–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, J.; Yasuda, H.; Nonaka, Y.; Fujiwara, M.; Nakamura, Y.; Soejima, K.; Betsuyaku, T. The FGF2 aptamer inhibits the growth of FGF2-FGFR pathway driven lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 1330–1334. [Google Scholar] [CrossRef]
- Chévrier, M.; Brakch, N.; Lesueur, C.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerrière, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Krokhin, O.V.; Beavis, R.C.; Ries, M.; Robinson, C.; Goldin, E.; Brady, R.O.; Wilkins, J.A.; Schiffmann, R. Proteomics of specific treatment-related alterations in Fabry disease: A strategy to identify biological abnormalities. Proc. Natl. Acad. Sci. USA 2007, 104, 2873–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, M.A.; Désormeaux, A.; Labelle, A.; Soulez, M.; Soulez, G.; Langelier, Y.; Pshezhetsky, A.V.; Hébert, M.J. Endothelial stress induces the release of vitamin D-binding protein, a novel growth factor. Biochem. Biophys. Res. Commun. 2005, 338, 1374–1382. [Google Scholar] [CrossRef]
- Coughlin, P.B. Antiplasmin: The forgotten serpin? FEBS J. 2005, 272, 4852–4857. [Google Scholar] [CrossRef]
- Carey, M.J.; Rodgers, G.M. Disseminated Intravascular Coagulation: Clinical and Laboratory Aspects. Am. J. Hematol. 1998, 59, 65–73. [Google Scholar] [CrossRef]
- Hollander, Z.; Dai, D.L.Y.; Putko, B.N.; Yogasundaram, H.; Wilson-McManus, J.E.; Thompson, R.B.; Khan, A.; West, M.L.; McManus, B.M.; Oudit, G.Y. Gender-specific plasma proteomic biomarkers in patients with Anderson-Fabry disease. Eur. J. Heart Fail. 2015, 17, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doykov, I.D.; Heywood, W.E.; Nikolaenko, V.; Śpiewak, J.; Hällqvist, J.; Clayton, P.T.; Mills, P.; Warnock, D.G.; Nowak, A.; Mills, K. Rapid, proteomic urine assay for monitoring progressive organ disease in Fabry disease. J. Med. Genet. 2020, 57, 38–47. [Google Scholar] [CrossRef]
- Kistler, A.D.; Siwy, J.; Breunig, F.; Jeevaratnam, P.; Scherl, A.; Mullen, W.; Warnock, D.G.; Wanner, C.; Hughes, D.A.; Mischak, H.; et al. A distinct urinary biomarker pattern characteristic of female fabry patients that mirrors response to enzyme replacement therapy. PLoS ONE 2011, 6, e20534. [Google Scholar] [CrossRef]
- Manwaring, V.; Heywood, W.E.; Clayton, R.; Lachmann, R.H.; Keutzer, J.; Hindmarsh, P.; Winchester, B.; Heales, S.; Mills, K. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: Evidence of presymptomatic kidney disease in pediatric fabry and type-I diabetic patients. J. Proteome Res. 2013, 12, 2013–2021. [Google Scholar] [CrossRef]
- Vojtová, L.; Zima, T.; Tesař, V.; Michalová, J.; Přikryl, P.; Dostálová, G.; Linhart, A. Study of urinary proteomes in Anderson-Fabry disease. Ren. Fail. 2010, 32, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.T.M.; Kirsztajn, G.M.; Pereira, E.M.; Andrade, H.M.; Oliveira, I.H.R.; Labilloy, A.; da Silva, A.S. Proteomic profiling of engineered human immortalized podocyte cell model of Fabry disease. Mol. Genet. Metab. 2019, 126, S106–S107. [Google Scholar] [CrossRef]
- Slaats, G.G.; Braun, F.; Hoehne, M.; Frech, L.E.; Blomberg, L.; Benzing, T.; Schermer, B.; Rinschen, M.M.; Kurschat, C.E. Urine-derived cells: A promising diagnostic tool in Fabry disease patients. Sci. Rep. 2018, 8, 11042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’Imperio, V.; Smith, A.; Chinello, C.; Pagni, F.; Magni, F. Proteomics and glomerulonephritis: A complementary approach in renal pathology for the identification of chronic kidney disease related markers. Proteom. –Clin. Appl. 2016, 10, 371–383. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study | Method | Analysis | Results | Conclusion |
|---|---|---|---|---|
| Kistler et al. [40] | Mass spectrometry-based proteomics | Urinary proteomic biomarkers | Identified over 200 peptides associated with ADPKD; alteration of urinary collagen fragments; upregulation of fibrinogen alpha chain and of keratin; downregulation of c-terminal fragments of uromodulin; increased osteopontin fragments in the urine. | Urinary proteomic biomarkers can improve ADPKD diagnosis and risk stratification for better patient outcomes. |
| Pejchinovski et al. [47] | Mass spectrometry-based proteomics | Urine peptidome analysis | Identified 20 urinary peptidome biomarkers for predicting end-stage renal disease and ADPKD progression. The biomarker score was equivalent to that of the htTKV. Identified proteolytic pathways involved in ADPKD progression, which could serve as potential targets for therapeutic intervention. | Non-invasive diagnostic tool using urinary biomarkers can predict ADPKD progression and identify targets for therapy. |
| Rauniyar et al. [49] | Tandem mass tag-based proteomics | Quantification of protein expression | Identified potential urinary protein biomarkers of the cyst growth rate in ADPKD. | Urinary biomarkers could serve as non-invasive tools for ADPKD diagnosis and monitoring. |
| Salih et al. [54] | Mass spectrometry-based proteomics | Proteomic analysis of urinary extracellular vesicles (uEVs) | Identified potential biomarkers (plakins and complement proteins) and therapeutic targets for ADPKD. | The study provides insight into ADPKD progression and identifies potential targets for therapeutic intervention. |
| Pocsfalvi G. [53] | Mass spectrometry-based proteomics | EVs isolated from pooled urine samples | Identified 83 differentially expressed extracellular vesicle (EV) proteins involved in signal transduction pathways of primary cilia, Ca(2+)-activated signaling, cell-cycle regulation, and cell differentiation. The reduced levels of AQP-2 and increased levels of APO-A1 indicate impaired renal concentrating capability and may correlate with the decline in eGFR. | Quantitative proteomics of urinary EVs can be a useful tool in studying ADPKD. |
| Hogan et al. [50] | Electron microscopy and immunoblotting | Characterization of exosome-like vesicles | Identified 552 proteins implicated in signaling; confirmed the cleavage of polycystin-1 and fibrocystin. | Isolation from urine could be a non-invasive method for the diagnosis and monitoring of the disease. The study of PKD-ELVs and their relationship with primary cilia adds a novel aspect to our understanding of polycystic kidney. |
| Hogan et al. [51] | Mass spectrometry-based proteomics of urinary exosomes | Identification of biomarkers for PKD1 using urinary exosomes | Identified potential biomarkers for ADPKD using urinary exosomes; low PC1/TMEM2 ratios from the start of ADPKD; this ratio may have an inverse relationship with the htTKV. | Urinary exosomal biomarkers may have a clinical utility in the management of ADPKD. |
| Authors | Sample | Methods | Number of Patients | Results | Conclusion |
|---|---|---|---|---|---|
| Blood derived proteins | |||||
| Heo et al. [63] | Blood (before and after ERT) | 2D electrophoresis, MALDI-TOF MS, MS/MS. | Eight patients with classical FD. | Pre-ERT significantly increased:
| C3-mediated complement activation is changed in FD. ERT could promote its stabilisation. |
| Cigna et al. [67] | Blood (PBMC from FD patients) | 2D electrophoresis, MALDI-TOF MS. | Eight FD patients (30–59 years, 6 males and 2 females; 2 patients on ERT) and six healthy controls. | Downregulated proteins:
| Patients with FD display changes in the PMBC proteome compared to healthy subjects. |
| Moore et al. [75] | Blood (serum before and after 6 months of ERT) | O-methylisourea-based differential isotope labeling with tandem MS (MS/MS) | Thirteen children (6.5–17 years) | Decrease after ERT:
| Present abnormalities of angiogenesis factors and fibrinolysis. |
| Hollander et al. [79] | Blood | LC-MS/MS, iTRAQ | Thirty-two FD patients; 14 healthy controls. | Proteins sensitive and specific for male patients:
| Gender-specific plasma protein biomarker panels were identified. |
| Urine derived proteins | |||||
| Matafora et al. [66] | Urine | LC-MS/MS | Eleven FD patients non-ERT treated and twelve ERT-treated patients; twelve healthy controls. | Upregulated proteins:
| The urinary proteome of FD patients is different from healthy controls; upregulated proteins are decreased after ERT. |
| Doykov et al. [80] | Urine | LC-MS/MS | Sixty-six patients (27 males, 39 females) | Urinary proteins elevated in the early stage/asymptomatic patients:
| Glycogen phosphorylase brain form was the only protein elevated from the early-stage and continued to increase with progressive multiorgan involvement. Protein biomarkers might be used for the monitoring of therapy or disease progression. |
| Kistler et al. [81] | Urine | CE-MS; Micro-TOF MS | Thirty-five FD female patients (non-treated); eighty-nine healthy controls. | Sixty-four identified diagnostic biomarkers (88.2% sensitivity, 97.8% specificity) | Urinary biomarker model performing well in diagnosis of FD in female patients and in monitoring the response to ERT. |
| Manwaring et al. [82] | Urine | LC-MS/MS | Ten pediatric FD male patients; 6–16 years. | Prosaposin and GM2AP were elevated in FD patients and reduced after 12 months of ERT. | Protein biomarkers could be used for monitoring the response to ERT. |
| Vojtová et al. [83] | Urine (comparison between FD patients and healthy controls) | 2D electrophoresis images of urine samples, MALDI-TOF MS | Twenty FD patients (18–69 years; 11 males, 9 females), 13 patients were on ERT, Ten control subjects (27–42 years; 5 males, 5 females). | Abundant proteins in FD patients were:
| No significant qualitative differences between treated and untreated FD patients. Molecular size of H2 d-isomerase was modified. |
| Proteins derived from cell model | |||||
| Neto et al. [84] | Cell model (human podocytes) | Two-dimensional differential gel electrophoresis (2D-DIGE), MALDI-TOF MS. | Not applicable. | Downregulated proteins:
| FD podocytes express a profibrotic proliferative pattern. |
| Slaats et al. [85] | Urine-derived cells | nLC-MS/MS | Seven patients (5 males and 2 females; 26–68 years) | Increased proteins:
| Urine-derived cells from FD patients could be used as diagnostic tools, ERT monitoring, and testing therapeutic interventions. |
| Renal biopsy | |||||
| L’Imperio et al. [86] | Renal biopsy | MALDI-TOF MS, MALDI MS/MS, MALDI-MSI. | Fourteen FD patients (6 males, 8 females, 19–66 years) | Differences in protein expression between female and male FD patients, as well as between classic and atypical variants. | MALDI-MSI allows for phenotypic distinction in FD and possibility of genetic classification. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rroji, M.; Figurek, A.; Spasovski, G. Proteomic Approaches and Potential Applications in Autosomal Dominant Polycystic Kidney Disease and Fabry Disease. Diagnostics 2023, 13, 1152. https://doi.org/10.3390/diagnostics13061152
Rroji M, Figurek A, Spasovski G. Proteomic Approaches and Potential Applications in Autosomal Dominant Polycystic Kidney Disease and Fabry Disease. Diagnostics. 2023; 13(6):1152. https://doi.org/10.3390/diagnostics13061152
Chicago/Turabian StyleRroji, Merita, Andreja Figurek, and Goce Spasovski. 2023. "Proteomic Approaches and Potential Applications in Autosomal Dominant Polycystic Kidney Disease and Fabry Disease" Diagnostics 13, no. 6: 1152. https://doi.org/10.3390/diagnostics13061152