
Clinical Improvement in Depression and Cognitive Deficit Following Electroconvulsive Therapy


Abstract
1. Introduction
2. ECT Alters the Brain Structures
2.1. Structural Changes in the Hippocampus in Depression and Following ECT
2.2. Structural Changes in the Amygdala in Depression and Following ECT
2.3. Structural Changes in Other Brain Regions in Depression and Following ECT
2.4. Influence of Electrode Placement and Number of ECT Sessions on Brain Structural Changes
2.5. The Relationship between Post-ECT Volume Changes and Clinical Outcome
2.6. The Relationship between Post-ECT Volume Changes and Cognitive Outcome
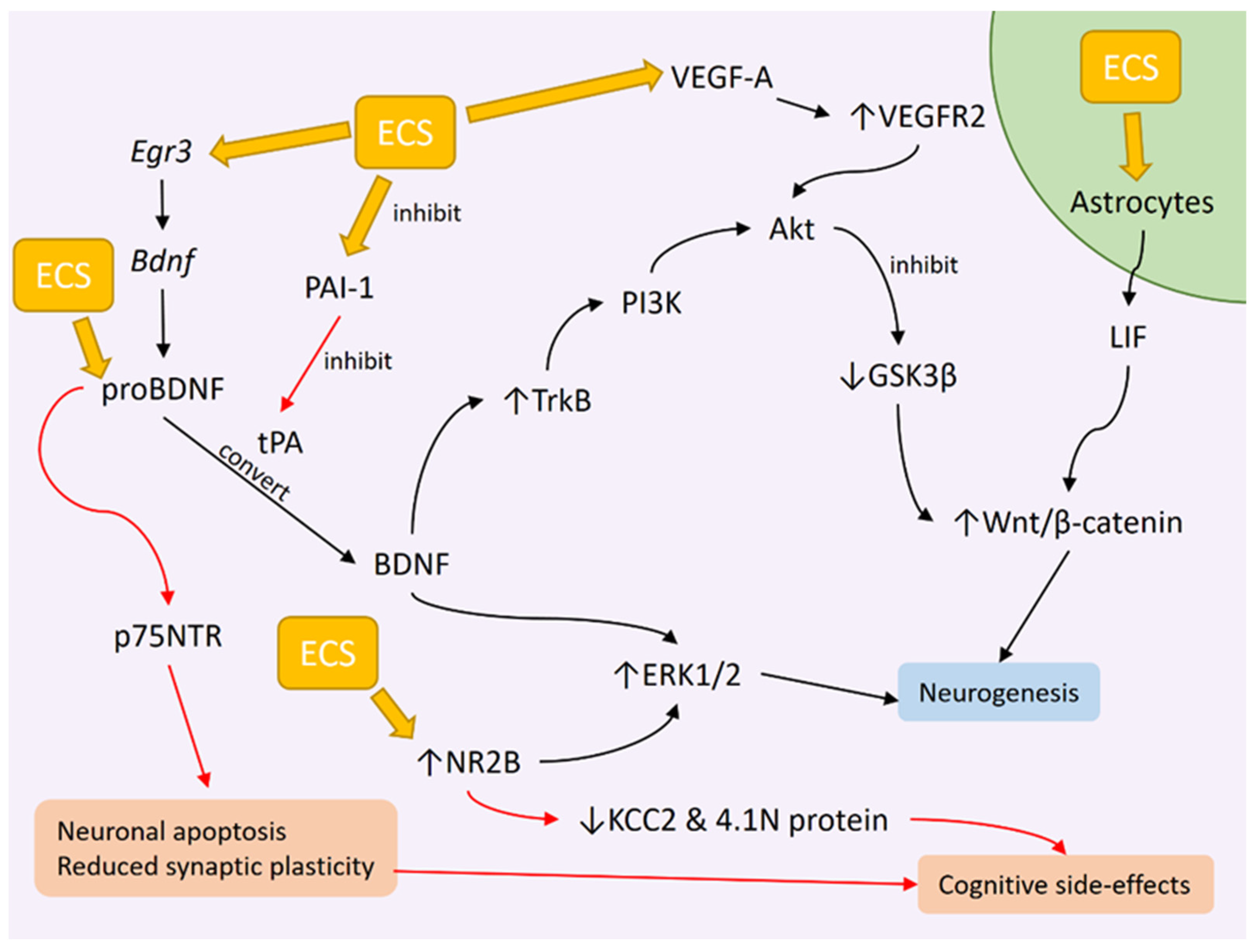
3. ECT Induces Neurogenesis
3.1. ECS Promotes Hippocampal Neurogenesis in Animals
3.2. ECT Effects on BDNF Levels
3.3. Genetic Changes Influencing BDNF Expression as a Result of ECS
3.4. ECT Effects on BDNF Downstream Signaling
3.5. Potential Role of proBDNF in Disrupting Cognitive Function Following ECT
4. VEGF Changes after ECT
5. ECT and Cerebral Edema
5.1. Compromised Neuronal Integrity Post-ECT
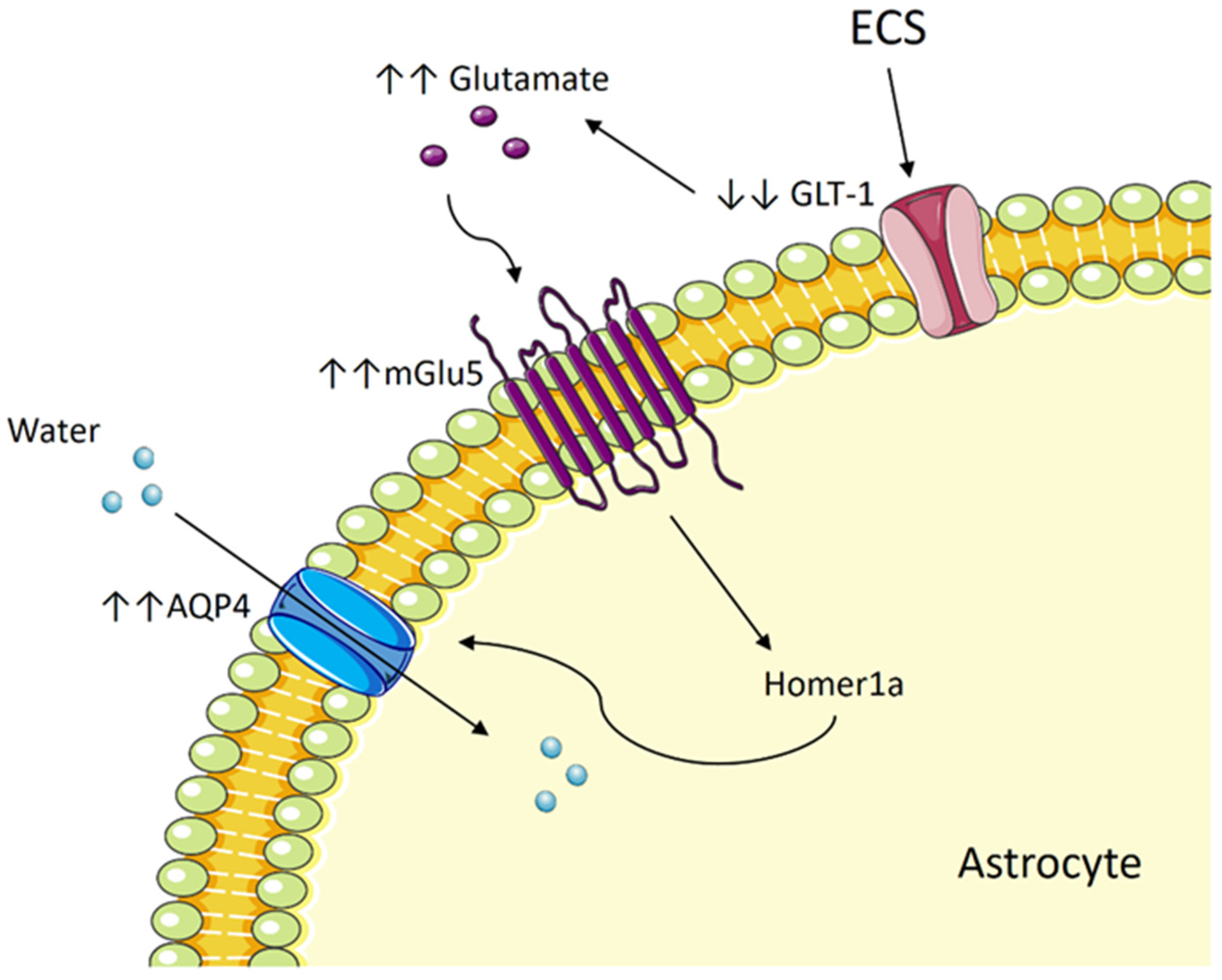
5.2. Could ECT Result in Cerebral Edema?
5.3. ECT Induces Cytotoxic Edema in Astrocytes by Way of Aquaporin and Glutamate
5.4. The Potential Role of mGlu5 and Homer1a
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- König, H.; König, H.H.; Konnopka, A. The excess costs of depression: A systematic review and meta-analysis. Epidemiol. Psychiatr. Sci. 2019, 29, e30. [Google Scholar] [CrossRef]
- Diagnostic and Statistical Manual of Mental Disorders DSM-IV, 4th ed.; American Psychiatric Association: Washington, DC, USA, 1994.
- Enev, M.; McNally, K.A.; Varghese, G.; Zubal, I.G.; Ostroff, R.B.; Blumenfeld, H. Imaging Onset and Propagation of ECT-induced Seizures. Epilepsia 2007, 48, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Lin, C.H.; Chiu, Y.C.; Tseng, C.C. The Clonic Phase of Seizures in Patients Treated with Electroconvulsive Therapy is Related to Age and Stimulus Intensity. Front. Psychiatry 2013, 4, 166. [Google Scholar] [CrossRef]
- Ray, A.K. Does electroconvulsive therapy cause epilepsy? J. ECT 2013, 29, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.L.; Zivin, K.; Maixner, D.F. Cost-effectiveness of Electroconvulsive Therapy vs Pharmacotherapy/Psychotherapy for Treatment-Resistant Depression in the United States. JAMA Psychiatry 2018, 75, 713–722. [Google Scholar] [CrossRef]
- Elias, A.; Phutane, V.H.; Clarke, S.; Prudic, J. Electroconvulsive therapy in the continuation and maintenance treatment of depression: Systematic review and meta-analyses. Aust. N. Z. J. Psychiatry 2018, 52, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Hermida, A.P.; Glass, O.M.; Shafi, H.; McDonald, W.M. Electroconvulsive Therapy in Depression: Current Practice and Future Direction. Psychiatr. Clin. N. Am. 2018, 41, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Venkatasalam, J.; Suzaily, W. Agitated Depression: The Challenges in Management and the Role of Maintenance Electroconvulsive Therapy. IIUM Med. J. Malays. 2019, 18, 166–168. [Google Scholar] [CrossRef]
- Park, M.J.; Kim, H.; Kim, E.J.; Yook, V.; Chung, I.W.; Lee, S.M.; Jeon, H.J. Recent Updates on Electro-Convulsive Therapy in Patients with Depression. Psychiatry Investig. 2021, 18, 1–10. [Google Scholar] [CrossRef]
- van Diermen, L.; van den Ameele, S.; Kamperman, A.M.; Sabbe, B.C.G.; Vermeulen, T.; Schrijvers, D.; Birkenhäger, T.K. Prediction of electroconvulsive therapy response and remission in major depression: Meta-analysis. Br. J. Psychiatry J. Ment. Sci. 2018, 212, 71–80. [Google Scholar] [CrossRef]
- Weiss, A.; Hussain, S.; Ng, B.; Sarma, S.; Tiller, J.; Waite, S.; Loo, C. Royal Australian and New Zealand College of Psychiatrists professional practice guidelines for the administration of electroconvulsive therapy. Aust. N. Z. J. Psychiatry 2019, 53, 609–623. [Google Scholar] [CrossRef]
- Dong, M.; Zhu, X.M.; Zheng, W.; Li, X.H.; Ng, C.H.; Ungvari, G.S.; Xiang, Y.T. Electroconvulsive therapy for older adult patients with major depressive disorder: A systematic review of randomized controlled trials. Psychogeriatrics 2018, 18, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Zhao, L.B.; Liu, Y.Y.; Fan, S.H.; Xie, P. Comparative efficacy and acceptability of electroconvulsive therapy versus repetitive transcranial magnetic stimulation for major depression: A systematic review and multiple-treatments meta-analysis. Behav. Brain Res. 2017, 320, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.; Arumugham, S.S.; Thirthalli, J. Adverse Effects of Electroconvulsive Therapy. Psychiatr. Clin. N. Am. 2016, 39, 513–530. [Google Scholar] [CrossRef]
- Gbyl, K.; Stottrup, M.M.; Raghava, J.M.; Jie, S.X.; Videbech, P. Hippocampal volume and memory impairment after electroconvulsive therapy in patients with depression. Acta Psychiatr. Scand. 2021, 143, 238–252. [Google Scholar] [CrossRef]
- Ramlan, H.; Shafri, N.I.; Wahab, S.; Kamarudin, M.A.; Rajikan, R.; Abdul Wahab, N.A.; Damanhuri, H.A. Depression, Anxiety and Stress in Medical Students: An Early Observation Analysis. Mediterr. J. Clin. Psychol. 2020, 8, 1–16. [Google Scholar] [CrossRef]
- van Buel, E.M.; Patas, K.; Peters, M.; Bosker, F.J.; Eisel, U.L.M.; Klein, H.C. Immune and neurotrophin stimulation by electroconvulsive therapy: Is some inflammation needed after all? Transl. Psychiatry 2015, 5, e609. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.L.; Swartz, H.A. A Critical Appraisal of Neuroimaging Studies of Bipolar Disorder: Toward a New Conceptualization of Underlying Neural Circuitry and a Road Map for Future Research. Am. J. Psychiatry 2014, 171, 829–843. [Google Scholar] [CrossRef]
- Bai, T.; Wei, Q.; Xie, W.; Wang, A.; Wang, J.; Ji, G.J.; Wang, K.; Tian, Y. Hippocampal-subregion functional alterations associated with antidepressant effects and cognitive impairments of electroconvulsive therapy. Psychol. Med. 2019, 49, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, D.M.; Sprengel, R.; Sanderson, D.J.; McHugh, S.B.; Rawlins, J.N.P.; Monyer, H.; Seeburg, P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 2014, 15, 181–192. [Google Scholar] [CrossRef]
- Barkus, C.; McHugh, S.B.; Sprengel, R.; Seeburg, P.H.; Rawlins, J.N.P.; Bannerman, D.M. Hippocampal NMDA receptors and anxiety: At the interface between cognition and emotion. Eur. J. Pharmacol. 2010, 626, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Nawa, N.E.; Ando, H. Effective connectivity within the ventromedial prefrontal cortex-hippocampus-amygdala network during the elaboration of emotional autobiographical memories. NeuroImage 2019, 189, 316–328. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef]
- Hei, M.; Chen, P.; Wang, S.; Li, X.; Xu, M.; Zhu, X.; Wang, Y.; Duan, J.; Huang, Y.; Zhao, S. Effects of chronic mild stress induced depression on synaptic plasticity in mouse hippocampus. Behav. Brain Res. 2019, 365, 26–35. [Google Scholar] [CrossRef]
- Kandilarova, S.; Stoyanov, D.; Kostianev, S.; Specht, K. Altered Resting State Effective Connectivity of Anterior Insula in Depression. Front. Psychiatry 2018, 9, 83. [Google Scholar] [CrossRef]
- Picó-Pérez, M.; Radua, J.; Steward, T.; Menchón, J.M.; Soriano-Mas, C. Emotion regulation in mood and anxiety disorders: A meta-analysis of fMRI cognitive reappraisal studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, F.; Wolf, N.D.; Pennuto, M.; Vasic, N.; Wolf, R.C. Revisiting default mode network function in major depression: Evidence for disrupted subsystem connectivity. Psychol. Med. 2014, 44, 2041–2051. [Google Scholar] [CrossRef]
- Whitfield-Gabrieli, S.; Ford, J.M. Default Mode Network Activity and Connectivity in Psychopathology. Annu. Rev. Clin. Psychol. 2012, 8, 49–76. [Google Scholar] [CrossRef]
- Cavaleri, D.; Bartoli, F. Biomolecular Research on Electroconvulsive Therapy for Mental Disorders: State of the Art and Future Directions. Alpha Psychiatry 2022, 23, 57–58. [Google Scholar] [CrossRef]
- Moica, T.; Gligor, A.; Moica, S. The Relationship between Cortisol and the Hippocampal Volume in Depressed Patients—A MRI Pilot Study. Procedia Technol. 2016, 22, 1106–1112. [Google Scholar] [CrossRef]
- Merz, E.C.; Desai, P.M.; Maskus, E.A.; Melvin, S.A.; Rehman, R.; Torres, S.D.; Meyer, J.; He, X.; Noble, K.G. Socioeconomic Disparities in Chronic Physiologic Stress Are Associated With Brain Structure in Children. Biol. Psychiatry 2019, 86, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, L.R.; Noble, K.G. Perceived stress is associated with smaller hippocampal volume in adolescence. Psychophysiology 2018, 55, e13025. [Google Scholar] [CrossRef] [PubMed]
- Barch, D.M.; Harms, M.P.; Tillman, R.; Hawkey, E.; Luby, J.L. Early childhood depression, emotion regulation, episodic memory, and hippocampal development. J. Abnorm. Psychol. 2019, 128, 81–95. [Google Scholar] [CrossRef]
- Barch, D.M.; Tillman, R.; Kelly, D.; Whalen, D.; Gilbert, K.; Luby, J.L. Hippocampal volume and depression among young children. Psychiatry Res. Neuroimaging 2019, 288, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Potter, G.G.; McQuoid, D.R.; Boyd, B.; Turner, R.; MacFall, J.R.; Taylor, W.D. Effects of early life stress on depression, cognitive performance and brain morphology. Psychol. Med. 2017, 47, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Tannous, J.; Godlewska, B.R.; Tirumalaraju, V.; Soares, J.C.; Cowen, P.J.; Selvaraj, S. Stress, inflammation and hippocampal subfields in depression: A 7 Tesla MRI Study. Transl. Psychiatry 2020, 10, 78. [Google Scholar] [CrossRef]
- Schoenfeld, T.J.; McCausland, H.C.; Morris, H.D.; Padmanaban, V.; Cameron, H.A. Stress and Loss of Adult Neurogenesis Differentially Reduce Hippocampal Volume. Biol. Psychiatry 2017, 82, 914–923. [Google Scholar] [CrossRef]
- Schmaal, L.; Veltman, D.J.; van Erp, T.G.; Sämann, P.G.; Frodl, T.; Jahanshad, N.; Loehrer, E.; Tiemeier, H.; Hofman, A.; Niessen, W.J.; et al. Subcortical brain alterations in major depressive disorder: Findings from the ENIGMA Major Depressive Disorder working group. Mol. Psychiatry 2016, 21, 806–812. [Google Scholar] [CrossRef]
- Jamieson, A.; Goodwill, A.M.; Termine, M.; Campbell, S.; Szoeke, C. Depression related cerebral pathology and its relationship with cognitive functioning: A systematic review. J. Affect. Disord. 2019, 250, 410–418. [Google Scholar] [CrossRef]
- Santos, M.A.O.; Bezerra, L.S.; Carvalho, A.; Brainer-Lima, A.M. Global hippocampal atrophy in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Trends Psychiatry Psychother. 2018, 40, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Wise, T.; Radua, J.; Via, E.; Cardoner, N.; Abe, O.; Adams, T.M.; Amico, F.; Cheng, Y.; Cole, J.H.; de Azevedo Marques Périco, C.; et al. Common and distinct patterns of grey-matter volume alteration in major depression and bipolar disorder: Evidence from voxel-based meta-analysis. Mol. Psychiatry 2017, 22, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Roddy, D.W.; Farrell, C.; Doolin, K.; Roman, E.; Tozzi, L.; Frodl, T.; O’Keane, V.; O’Hanlon, E. The Hippocampus in Depression: More Than the Sum of Its Parts? Advanced Hippocampal Substructure Segmentation in Depression. Biol. Psychiatry 2019, 85, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Zhang, Y.; Yang, Z.; Han, S.; Cheng, J. Reduced Brain Gray Matter Volume in Patients With First-Episode Major Depressive Disorder: A Quantitative Meta-Analysis. Front. Psychiatry 2021, 12, 671348. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Lippard, E.T.C.; Sankar, A.; Wallace, A.; Johnston, J.A.Y.; Wang, F.; Pittman, B.; Spencer, L.; Oquendo, M.A.; Blumberg, H.P. Gray and white matter differences in adolescents and young adults with prior suicide attempts across bipolar and major depressive disorders. J. Affect. Disord. 2019, 245, 1089–1097. [Google Scholar] [CrossRef]
- Maller, J.J.; Broadhouse, K.; Rush, A.J.; Gordon, E.; Koslow, S.; Grieve, S.M. Increased hippocampal tail volume predicts depression status and remission to anti-depressant medications in major depression. Mol. Psychiatry 2018, 23, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Noda, T.; Sato, N.; Okazaki, M.; Ishikawa, M.; Hattori, K.; Hori, H.; Sasayama, D.; Teraishi, T.; Sone, D.; et al. Effect of electroconvulsive therapy on gray matter volume in major depressive disorder. J. Affect. Disord. 2015, 186, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, A.; Demirakca, T.; Böhringer, A.; Clemm von Hohenberg, C.; Aksay, S.S.; Bumb, J.M.; Kranaster, L.; Ende, G. Electroconvulsive therapy increases temporal gray matter volume and cortical thickness. Eur. Neuropsychopharmacol. 2016, 26, 506–517. [Google Scholar] [CrossRef]
- Sartorius, A.; Demirakca, T.; Böhringer, A.; Clemm von Hohenberg, C.; Aksay, S.S.; Bumb, J.M.; Kranaster, L.; Nickl-Jockschat, T.; Grözinger, M.; Thomann, P.A.; et al. Electroconvulsive therapy induced gray matter increase is not necessarily correlated with clinical data in depressed patients. Brain Stimul. 2019, 12, 335–343. [Google Scholar] [CrossRef]
- Camilleri, J.A.; Hoffstaedter, F.; Zavorotny, M.; Zöllner, R.; Wolf, R.C.; Thomann, P.; Redlich, R.; Opel, N.; Dannlowski, U.; Grözinger, M.; et al. Electroconvulsive therapy modulates grey matter increase in a hub of an affect processing network. NeuroImage Clin. 2020, 25, 102114. [Google Scholar] [CrossRef]
- Wilkinson, S.T.; Sanacora, G.; Bloch, M.H. Hippocampal Volume Changes Following Electroconvulsive Therapy: A Systematic Review and Meta-analysis. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 327–335. [Google Scholar] [CrossRef]
- Gbyl, K.; Rostrup, E.; Raghava, J.M.; Andersen, C.; Rosenberg, R.; Larsson, H.B.W.; Videbech, P. Volume of hippocampal subregions and clinical improvement following electroconvulsive therapy in patients with depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104, 110048. [Google Scholar] [CrossRef]
- Cao, B.; Luo, Q.; Fu, Y.; Du, L.; Qiu, T.; Yang, X.; Chen, X.; Chen, Q.; Soares, J.C.; Cho, R.Y.; et al. Predicting individual responses to the electroconvulsive therapy with hippocampal subfield volumes in major depression disorder. Sci. Rep. 2018, 8, 5434. [Google Scholar] [CrossRef] [PubMed]
- Gryglewski, G.; Baldinger-Melich, P.; Seiger, R.; Godbersen, G.M.; Michenthaler, P.; Klobl, M.; Spurny, B.; Kautzky, A.; Vanicek, T.; Kasper, S.; et al. Structural changes in amygdala nuclei, hippocampal subfields and cortical thickness following electroconvulsive therapy in treatment-resistant depression: Longitudinal analysis. Br. J. Psychiatry 2019, 214, 159–167. [Google Scholar] [CrossRef]
- Takamiya, A.; Plitman, E.; Chung, J.K.; Chakravarty, M.; Graff-Guerrero, A.; Mimura, M.; Kishimoto, T. Acute and long-term effects of electroconvulsive therapy on human dentate gyrus. Neuropsychopharmacology 2019, 44, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Kubicki, A.; Leaver, A.M.; Vasavada, M.; Njau, S.; Wade, B.; Joshi, S.H.; Loureiro, J.; Hellemann, G.; Woods, R.P.; Espinoza, R.; et al. Variations in Hippocampal White Matter Diffusivity Differentiate Response to Electroconvulsive Therapy in Major Depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 300–309. [Google Scholar] [CrossRef]
- Nuninga, J.O.; Mandl, R.C.W.; Boks, M.P.; Bakker, S.; Somers, M.; Heringa, S.M.; Nieuwdorp, W.; Hoogduin, H.; Kahn, R.S.; Luijten, P.; et al. Volume increase in the dentate gyrus after electroconvulsive therapy in depressed patients as measured with 7T. Mol. Psychiatry 2020, 25, 1559–1568. [Google Scholar] [CrossRef]
- Bartsch, T.; Döhring, J.; Rohr, A.; Jansen, O.; Deuschl, G. CA1 neurons in the human hippocampus are critical for autobiographical memory, mental time travel, and autonoetic consciousness. Proc. Natl. Acad. Sci. USA 2011, 108, 17562. [Google Scholar] [CrossRef]
- Kumar, J.; Hapidin, H.; Get Bee, Y.T.; Ismail, Z. The effects of acute ethanol administration on ethanol withdrawal-induced anxiety-like syndrome in rats: A biochemical study. Alcohol 2016, 50, 9–17. [Google Scholar] [CrossRef]
- Sandu, A.L.; Artiges, E.; Galinowski, A.; Gallarda, T.; Bellivier, F.; Lemaitre, H.; Granger, B.; Ringuenet, D.; Tzavara, E.T.; Martinot, J.L.; et al. Amygdala and regional volumes in treatment-resistant versus nontreatment-resistant depression patients. Depress. Anxiety 2017, 34, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Espinoza Oyarce, D.A.; Shaw, M.E.; Alateeq, K.; Cherbuin, N. Volumetric brain differences in clinical depression in association with anxiety: A systematic review with meta-analysis. J. Psychiatry Neurosci. JPN 2020, 45, 406–429. [Google Scholar] [CrossRef]
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef]
- Zhang, W.-H.; Liu, W.-Z.; He, Y.; You, W.-J.; Zhang, J.-Y.; Xu, H.; Tian, X.-L.; Li, B.-M.; Mei, L.; Holmes, A.; et al. Chronic Stress Causes Projection-Specific Adaptation of Amygdala Neurons via Small-Conductance Calcium-Activated Potassium Channel Downregulation. Biol. Psychiatry 2019, 85, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.H.; Espinoza, R.T.; Pirnia, T.; Shi, J.; Wang, Y.; Ayers, B.; Leaver, A.; Woods, R.P.; Narr, K.L. Structural Plasticity of the Hippocampus and Amygdala Induced by Electroconvulsive Therapy in Major Depression. Biol. Psychiatry 2016, 79, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Wei, Q.; Bai, T.J.; Zhou, X.Q.; Sun, H.; Becker, B.; Tian, Y.H.; Wang, K.; Kendrick, K. Electroconvulsive therapy selectively enhanced feedforward connectivity from fusiform face area to amygdala in major depressive disorder. Soc. Cogn. Affect. Neurosci. 2017, 12, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Damborská, A.; Honzírková, E.; Barteček, R.; Hořínková, J.; Fedorová, S.; Ondruš, Š.; Michel, C.M.; Rubega, M. Altered directed functional connectivity of the right amygdala in depression: High-density EEG study. Sci. Rep. 2020, 10, 4398. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, Y.; Chen, Z.; Long, J.; Dai, J.; Huang, X.; Lui, S.; Radua, J.; Vieta, E.; Kemp, G.J.; et al. Meta-analysis of cortical thickness abnormalities in medication-free patients with major depressive disorder. Neuropsychopharmacology 2020, 45, 703–712. [Google Scholar] [CrossRef]
- Schmaal, L.; Hibar, D.P.; Sämann, P.G.; Hall, G.B.; Baune, B.T.; Jahanshad, N.; Cheung, J.W.; van Erp, T.G.M.; Bos, D.; Ikram, M.A.; et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol. Psychiatry 2017, 22, 900–909. [Google Scholar] [CrossRef]
- Suh, J.S.; Schneider, M.A.; Minuzzi, L.; MacQueen, G.M.; Strother, S.C.; Kennedy, S.H.; Frey, B.N. Cortical thickness in major depressive disorder: A systematic review and meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 287–302. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Niu, C.; Zhong, S.; Hu, H.; Chen, P.; Zhang, S.; Chen, G.; Deng, F.; Lai, S.; et al. Common and distinct abnormal frontal-limbic system structural and functional patterns in patients with major depression and bipolar disorder. NeuroImage Clin. 2018, 20, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Aso, T.; Miyata, J.; Sugihara, G.; Hazama, M.; Nemoto, K.; Yoshihara, Y.; Matsumoto, Y.; Okada, T.; Togashi, K.; et al. Early and late effects of electroconvulsive therapy associated with different temporal lobe structures. Transl. Psychiatry 2020, 10, 344. [Google Scholar] [CrossRef]
- Mulders, P.C.R.; Llera, A.; Beckmann, C.F.; Vandenbulcke, M.; Stek, M.; Sienaert, P.; Redlich, R.; Petrides, G.; Oudega, M.L.; Oltedal, L.; et al. Structural changes induced by electroconvulsive therapy are associated with clinical outcome. Brain Stimul. 2020, 13, 696–704. [Google Scholar] [CrossRef]
- Bai, S.; Gálvez, V.; Dokos, S.; Martin, D.; Bikson, M.; Loo, C. Computational models of Bitemporal, Bifrontal and Right Unilateral ECT predict differential stimulation of brain regions associated with efficacy and cognitive side effects. Eur. Psychiatry 2017, 41, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Oltedal, L.; Narr, K.L.; Abbott, C.; Anand, A.; Argyelan, M.; Bartsch, H.; Dannlowski, U.; Dols, A.; van Eijndhoven, P.; Emsell, L.; et al. Volume of the Human Hippocampus and Clinical Response Following Electroconvulsive Therapy. Biol. Psychiatry 2018, 84, 574–581. [Google Scholar] [CrossRef]
- Cano, M.; Martinez-Zalacain, I.; Bernabeu-Sanz, A.; Contreras-Rodriguez, O.; Hernandez-Ribas, R.; Via, E.; de Arriba-Arnau, A.; Galvez, V.; Urretavizcaya, M.; Pujol, J.; et al. Brain volumetric and metabolic correlates of electroconvulsive therapy for treatment-resistant depression: A longitudinal neuroimaging study. Transl. Psychiatry 2017, 7, e1023. [Google Scholar] [CrossRef]
- Jehna, M.; Wurm, W.; Pinter, D.; Vogel, K.; Holl, A.; Hofmann, P.; Ebner, C.; Ropele, S.; Fuchs, G.; Kapfhammer, H.-P.; et al. Do increases in deep grey matter volumes after electroconvulsive therapy persist in patients with major depression? A longitudinal MRI-study. J. Affect. Disord. 2021, 281, 908–917. [Google Scholar] [CrossRef]
- Argyelan, M.; Oltedal, L.; Deng, Z.D.; Wade, B.; Bikson, M.; Joanlanne, A.; Sanghani, S.; Bartsch, H.; Cano, M.; Dale, A.M.; et al. Electric field causes volumetric changes in the human brain. Elife 2019, 8, e49115. [Google Scholar] [CrossRef] [PubMed]
- Ousdal, O.T.; Argyelan, M.; Narr, K.L.; Abbott, C.; Wade, B.; Vandenbulcke, M.; Urretavizcaya, M.; Tendolkar, I.; Takamiya, A.; Stek, M.L.; et al. Brain Changes Induced by Electroconvulsive Therapy Are Broadly Distributed. Biol. Psychiatry 2020, 87, 451–461. [Google Scholar] [CrossRef]
- Fridgeirsson, E.A.; Deng, Z.D.; Denys, D.; van Waarde, J.A.; van Wingen, G.A. Electric field strength induced by electroconvulsive therapy is associated with clinical outcome. NeuroImage Clin. 2021, 30, 102581. [Google Scholar] [CrossRef]
- Oltedal, L.; Bartsch, H.; Sørhaug, O.J.E.; Kessler, U.; Abbott, C.; Dols, A.; Stek, M.L.; Ersland, L.; Emsell, L.; van Eijndhoven, P.; et al. The Global ECT-MRI Research Collaboration (GEMRIC): Establishing a multi-site investigation of the neural mechanisms underlying response to electroconvulsive therapy. NeuroImage Clin. 2017, 14, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, A.; Chung, J.K.; Liang, K.C.; Graff-Guerrero, A.; Mimura, M.; Kishimoto, T. Effect of electroconvulsive therapy on hippocampal and amygdala volumes: Systematic review and meta-analysis. Br. J. Psychiatry J. Ment. Sci. 2018, 212, 19–26. [Google Scholar] [CrossRef]
- Xu, J.; Wang, J.; Bai, T.; Zhang, X.; Li, T.; Hu, Q.; Li, H.; Zhang, L.; Wei, Q.; Tian, Y.; et al. Electroconvulsive Therapy Induces Cortical Morphological Alterations in Major Depressive Disorder Revealed with Surface-Based Morphometry Analysis. Int. J. Neural Syst. 2019, 29, 1950005. [Google Scholar] [CrossRef]
- van Oostrom, I.; van Eijndhoven, P.; Butterbrod, E.; van Beek, M.H.; Janzing, J.; Donders, R.; Schene, A.; Tendolkar, I. Decreased Cognitive Functioning After Electroconvulsive Therapy Is Related to Increased Hippocampal Volume: Exploring the Role of Brain Plasticity. J. Ect. 2018, 34, 117–123. [Google Scholar] [CrossRef]
- Laroy, M.; Bouckaert, F.; Vansteelandt, K.; Ohhels, J.; Dols, A.; Emsell, L.; Stek, M.; Vandenhulcke, M.; Sienaert, P. Association between hippocampal volume change and change in memory following electroconvulsive therapy in late-life depression. Acta Psychiatr. Scand. 2019, 140, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Argyelan, M.; Lencz, T.; Kang, S.; Ali, S.; Masi, P.J.; Moyett, E.; Joanlanne, A.; Watson, P.; Sanghani, S.; Petrides, G.; et al. ECT-induced cognitive side effects are associated with hippocampal enlargement. Transl. Psychiatry 2021, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, M.; Berkers, R.; Morris, R.G.M.; Fernández, G. Angular Gyrus Involvement at Encoding and Retrieval Is Associated with Durable But Less Specific Memories. J. Neurosci. 2017, 37, 9474–9485. [Google Scholar] [CrossRef]
- Olesen, M.V.; Wörtwein, G.; Pakkenberg, B. Electroconvulsive stimulation, but not chronic restraint stress, causes structural alterations in adult rat hippocampus--a stereological study. Hippocampus 2015, 25, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Alemu, J.L.; Elberling, F.; Azam, B.; Pakkenberg, B.; Olesen, M.V. Electroconvulsive treatment prevents chronic restraint stress-induced atrophy of the hippocampal formation—A stereological study. Brain Behav. 2019, 9, e01195. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.V.; Wörtwein, G.; Folke, J.; Pakkenberg, B. Electroconvulsive stimulation results in long-term survival of newly generated hippocampal neurons in rats. Hippocampus 2017, 27, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Madsen, T.M.; Treschow, A.; Bengzon, J.; Bolwig, T.G.; Lindvall, O.; Tingström, A. Increased neurogenesis in a model of electroconvulsive therapy. Biol. Psychiatry 2000, 47, 1043–1049. [Google Scholar] [CrossRef]
- García-Cabrerizo, R.; Ledesma-Corvi, S.; Bis-Humbert, C.; García-Fuster, M.J. Sex differences in the antidepressant-like potential of repeated electroconvulsive seizures in adolescent and adult rats: Regulation of the early stages of hippocampal neurogenesis. Eur. Neuropsychopharmacol. 2020, 41, 132–145. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Kee, N.; Sivalingam, S.; Boonstra, R.; Wojtowicz, J.M. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J. Neurosci. Methods 2002, 115, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Ure, K.; Ables, J.L.; Lagace, D.C.; Nave, K.-A.; Goebbels, S.; Eisch, A.J.; Hsieh, J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 2009, 12, 1090–1092. [Google Scholar] [CrossRef]
- Ueno, M.; Sugimoto, M.; Ohtsubo, K.; Sakai, N.; Endo, A.; Shikano, K.; Imoto, Y.; Segi-Nishida, E. The effect of electroconvulsive seizure on survival, neuronal differentiation, and expression of the maturation marker in the adult mouse hippocampus. J. Neurochem. 2019, 149, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Bonde, S.; Ekdahl, C.T.; Lindvall, O. Long-term neuronal replacement in adult rat hippocampus after status epilepticus despite chronic inflammation. Eur. J. Neurosci. 2006, 23, 965–974. [Google Scholar] [CrossRef]
- Jakubs, K.; Nanobashvili, A.; Bonde, S.; Ekdahl, C.T.; Kokaia, Z.; Kokaia, M.; Lindvall, O. Environment matters: Synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron 2006, 52, 1047–1059. [Google Scholar] [CrossRef]
- Jackson, J.; Chugh, D.; Nilsson, P.; Wood, J.; Carlström, K.; Lindvall, O.; Ekdahl, C.T. Altered synaptic properties during integration of adult-born hippocampal neurons following a seizure insult. PLoS ONE 2012, 7, e35557. [Google Scholar] [CrossRef]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Şahin, T.D.; Karson, A.; Balcı, F.; Yazır, Y.; Bayramgürler, D.; Utkan, T. TNF-alpha inhibition prevents cognitive decline and maintains hippocampal BDNF levels in the unpredictable chronic mild stress rat model of depression. Behav. Brain Res. 2015, 292, 233–240. [Google Scholar] [CrossRef]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the pathophysiology and treatment of depression: Activity-dependent effects distinguish rapid-acting antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Pisoni, A.; Strawbridge, R.; Hodsoll, J.; Powell, T.R.; Breen, G.; Hatch, S.; Hotopf, M.; Young, A.H.; Cleare, A.J. Growth Factor Proteins and Treatment-Resistant Depression: A Place on the Path to Precision. Front. Psychiatry 2018, 9, 386. [Google Scholar] [CrossRef]
- Rapinesi, C.; Kotzalidis, G.D.; Curto, M.; Serata, D.; Ferri, V.R.; Scatena, P.; Carbonetti, P.; Napoletano, F.; Miele, J.; Scaccianoce, S.; et al. Electroconvulsive therapy improves clinical manifestations of treatment-resistant depression without changing serum BDNF levels. Psychiatry Res. 2015, 227, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, F.; Dols, A.; Emsell, L.; De Winter, F.L.; Vansteelandt, K.; Claes, L.; Sunaert, S.; Stek, M.; Sienaert, P.; Vandenbulcke, M. Relationship Between Hippocampal Volume, Serum BDNF, and Depression Severity Following Electroconvulsive Therapy in Late-Life Depression. Neuropsychopharmacology 2016, 41, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Cavaleri, D.; Moretti, F.; Bartoccetti, A.; Mauro, S.; Crocamo, C.; Carrà, G.; Bartoli, F. The role of BDNF in major depressive disorder, related clinical features, and antidepressant treatment: Insight from meta-analyses. Neurosci. Biobehav. Rev. 2023, 149, 105159. [Google Scholar] [CrossRef] [PubMed]
- Luan, S.; Zhou, B.; Wu, Q.; Wan, H.; Li, H. Brain-derived neurotrophic factor blood levels after electroconvulsive therapy in patients with major depressive disorder: A systematic review and meta-analysis. Asian J. Psychiatry 2020, 51, 101983. [Google Scholar] [CrossRef]
- Rocha, R.B.; Dondossola, E.R.; Grande, A.J.; Colonetti, T.; Ceretta, L.B.; Passos, I.C.; Quevedo, J.; da Rosa, M.I. Increased BDNF levels after electroconvulsive therapy in patients with major depressive disorder: A meta-analysis study. J. Psychiatr. Res. 2016, 83, 47–53. [Google Scholar] [CrossRef]
- Ryan, K.M.; Dunne, R.; McLoughlin, D.M. BDNF plasma levels and genotype in depression and the response to electroconvulsive therapy. Brain Stimul. 2018, 11, 1123–1131. [Google Scholar] [CrossRef]
- Vanicek, T.; Kranz, G.S.; Vyssoki, B.; Fugger, G.; Komorowski, A.; Höflich, A.; Saumer, G.; Milovic, S.; Lanzenberger, R.; Eckert, A.; et al. Acute and subsequent continuation electroconvulsive therapy elevates serum BDNF levels in patients with major depression. Brain Stimul. 2019, 12, 1041–1050. [Google Scholar] [CrossRef]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Effect of electroconvulsive therapy on brain-derived neurotrophic factor levels in patients with major depressive disorder. Brain Behav. 2018, 8, e01101. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Elfving, B.; Plougmann, P.H.; Wegener, G. Detection of brain-derived neurotrophic factor (BDNF) in rat blood and brain preparations using ELISA: Pitfalls and solutions. J. Neurosci. Methods 2010, 187, 73–77. [Google Scholar] [CrossRef]
- Arosio, B.; Guerini, F.R.; Voshaar, R.C.O.; Aprahamian, I. Blood Brain-Derived Neurotrophic Factor (BDNF) and Major Depression: Do We Have a Translational Perspective? Front. Behav. Neurosci. 2021, 15, 626906. [Google Scholar] [CrossRef]
- Jorgensen, A.; Magnusson, P.; Hanson, L.G.; Kirkegaard, T.; Benveniste, H.; Lee, H.; Svarer, C.; Mikkelsen, J.D.; Fink-Jensen, A.; Knudsen, G.M.; et al. Regional brain volumes, diffusivity, and metabolite changes after electroconvulsive therapy for severe depression. Acta Psychiatr. Scand. 2016, 133, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Freire, T.F.V.; de Almeida Fleck, M.P.; da Rocha, N.S. Remission of depression following electroconvulsive therapy (ECT) is associated with higher levels of brain-derived neurotrophic factor (BDNF). Brain Res. Bull. 2016, 121, 263–269. [Google Scholar] [CrossRef]
- Petrides, G.; Tobias, K.G.; Kellner, C.H.; Rudorfer, M.V. Continuation and maintenance electroconvulsive therapy for mood disorders: Review of the literature. Neuropsychobiology 2011, 64, 129–140. [Google Scholar] [CrossRef]
- Vanicek, T.; Kranz, G.S.; Vyssoki, B.; Komorowski, A.; Fugger, G.; Höflich, A.; Micskei, Z.; Milovic, S.; Lanzenberger, R.; Eckert, A.; et al. Repetitive enhancement of serum BDNF subsequent to continuation ECT. Acta Psychiatr. Scand. 2019, 140, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Mindt, S.; Neumaier, M.; Hellweg, R.; Sartorius, A.; Kranaster, L. Brain-Derived Neurotrophic Factor in the Cerebrospinal Fluid Increases During Electroconvulsive Therapy in Patients With Depression: A Preliminary Report. J. ECT 2020, 36, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Gadad, B.S.; Vargas-Medrano, J.; Ramos, E.I.; Najera, K.; Fagan, M.; Forero, A.; Thompson, P.M. Altered levels of interleukins and neurotrophic growth factors in mood disorders and suicidality: An analysis from periphery to central nervous system. Transl. Psychiatry 2021, 11, 341. [Google Scholar] [CrossRef]
- Chen, F.; Ardalan, M.; Elfving, B.; Wegener, G.; Madsen, T.M.; Nyengaard, J.R. Mitochondria Are Critical for BDNF-Mediated Synaptic and Vascular Plasticity of Hippocampus following Repeated Electroconvulsive Seizures. Int. J. Neuropsychopharmacol. 2018, 21, 291–304. [Google Scholar] [CrossRef]
- Luo, J.; Min, S.; Wei, K.; Cao, J.; Wang, B.; Li, P.; Dong, J.; Liu, Y. Behavioral and molecular responses to electroconvulsive shock differ between genetic and environmental rat models of depression. Psychiatry Res. 2015, 226, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Dyrvig, M.; Christiansen, S.H.; Woldbye, D.P.; Lichota, J. Temporal gene expression profile after acute electroconvulsive stimulation in the rat. Gene 2014, 539, 8–14. [Google Scholar] [CrossRef]
- Meyers, K.T.; Marballi, K.K.; Brunwasser, S.J.; Renda, B.; Charbel, M.; Marrone, D.F.; Gallitano, A.L. The Immediate Early Gene Egr3 Is Required for Hippocampal Induction of Bdnf by Electroconvulsive Stimulation. Front. Behav. Neurosci. 2018, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Mariga, A.; Glaser, J.; Mathias, L.; Xu, D.; Xiao, M.; Worley, P.; Ninan, I.; Chao, M.V. Definition of a Bidirectional Activity-Dependent Pathway Involving BDNF and Narp. Cell Rep. 2015, 13, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.D.; Vaidya, P.V.; Retzbach, E.P.; Chung, S.J.; Kim, U.; Baselice, K.; Maynard, K.; Stepanian, A.; Staley, M.; Xiao, L.; et al. Narp Mediates Antidepressant-Like Effects of Electroconvulsive Seizures. Neuropsychopharmacology 2018, 43, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.-H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB Regulates Hippocampal Neurogenesis and Governs Sensitivity to Antidepressive Treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef]
- Enomoto, S.; Shimizu, K.; Nibuya, M.; Suzuki, E.; Nagata, K.; Kondo, T. Activated brain-derived neurotrophic factor/TrkB signaling in rat dorsal and ventral hippocampi following 10-day electroconvulsive seizure treatment. Neurosci. Lett. 2017, 660, 45–50. [Google Scholar] [CrossRef]
- Rantamaki, T. TrkB neurotrophin receptor at the core of antidepressant effects, but how? Cell Tissue Res. 2019, 377, 115–124. [Google Scholar] [CrossRef]
- Kohtala, S.; Theilmann, W.; Rosenholm, M.; Penna, L.; Karabulut, G.; Uusitalo, S.; Järventausta, K.; Yli-Hankala, A.; Yalcin, I.; Matsui, N.; et al. Cortical Excitability and Activation of TrkB Signaling During Rebound Slow Oscillations Are Critical for Rapid Antidepressant Responses. Mol. Neurobiol. 2019, 56, 4163–4174. [Google Scholar] [CrossRef]
- Zheng, Q.; Liu, L.; Liu, H.; Zheng, H.; Sun, H.; Ji, J.; Sun, Y.; Yang, T.; Zhao, H.; Qi, F.; et al. The Bu Shen Yi Sui Formula Promotes Axonal Regeneration via Regulating the Neurotrophic Factor BDNF/TrkB and the Downstream PI3K/Akt Signaling Pathway. Front. Pharmacol. 2019, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Pan, F.; Chen, J.; Chen, C.E.; Xie, D.P.; Jiang, X.Z.; Guo, S.J.; Zhou, J. Neuroprotection by plumbagin involves BDNF-TrkB-PI3K/Akt and ERK1/2/JNK pathways in isoflurane-induced neonatal rats. J. Pharm. Pharmacol. 2017, 69, 896–906. [Google Scholar] [CrossRef]
- Ahmed, S.; Kwatra, M.; Gawali, B.; Panda, S.R.; Naidu, V.G.M. Potential role of TrkB agonist in neuronal survival by promoting CREB/BDNF and PI3K/Akt signaling in vitro and in vivo model of 3-nitropropionic acid (3-NP)-induced neuronal death. Apoptosis. Int. J. Program. Cell Death 2021, 26, 52–70. [Google Scholar] [CrossRef]
- Gururajan, A.; Naughton, M.E.; Scott, K.A.; O’Connor, R.M.; Moloney, G.; Clarke, G.; Dowling, J.; Walsh, A.; Ismail, F.; Shorten, G.; et al. MicroRNAs as biomarkers for major depression: A role for let-7b and let-7c. Transl. Psychiatry 2016, 6, e862. [Google Scholar] [CrossRef] [PubMed]
- Paslakis, G.; Blum, W.F.; Deuschle, M. Intranasal insulin-like growth factor I (IGF-I) as a plausible future treatment of depression. Med. Hypotheses 2012, 79, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/β-catenin signaling. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, S.B.; Valenzuela-Bezanilla, D.; Mardones, M.D.; Varela-Nallar, L. Role of Wnt Signaling in Adult Hippocampal Neurogenesis in Health and Disease. Front. Cell Dev. Biol. 2020, 8, 860. [Google Scholar] [CrossRef]
- Dai, J.; Pan, J.Y.; Liao, N.; Shi, J.; Zeng, Q.; Huang, L.; Chen, L.P. Influence of miR-155 on behaviors of depression mice through regulating Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1398–1407. [Google Scholar] [CrossRef]
- Yao, Z.H.; Kang, X.; Yang, L.; Niu, Y.; Lu, Y.; Nie, L. PBA regulates neurogenesis and cognition dysfunction after repeated electroconvulsive shock in a rat model. Psychiatry Res. 2015, 230, 331–340. [Google Scholar] [CrossRef]
- Wang, J.Q.; Mao, L. The ERK Pathway: Molecular Mechanisms and Treatment of Depression. Mol. Neurobiol. 2019, 56, 6197–6205. [Google Scholar] [CrossRef]
- Albert-Gascó, H.; Ros-Bernal, F.; Castillo-Gómez, E.; Olucha-Bordonau, F.E. MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes. Int. J. Mol. Sci. 2020, 21, 4417. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Xu, X.; He, Z.; Wang, S.; Zhao, L.; Qiu, J.; Wang, D.; Gong, Z.; Qiu, X.; Huang, H. Antidepressant-Like Effects and Cognitive Enhancement of Coadministration of Chaihu Shugan San and Fluoxetine: Dependent on the BDNF-ERK-CREB Signaling Pathway in the Hippocampus and Frontal Cortex. BioMed Res. Int. 2020, 2020, 2794263. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhuang, F.Z.; Qin, S.J.; Zhou, L.; Wang, Y.; Shen, Q.F.; Li, M.; Villarreal, M.; Benefield, L.; Gu, S.L.; et al. Dexmedetomidine protects against learning and memory impairments caused by electroconvulsive shock in depressed rats: Involvement of the NMDA receptor subunit 2B (NR2B)-ERK signaling pathway. Psychiatry Res. 2016, 243, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Rantamäki, T.P.; Larsen, M.H.; Woldbye, D.P.; Mikkelsen, J.D.; Castrén, E.H. Rapid activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway by electroconvulsive shock in the rat prefrontal cortex is not associated with TrkB neurotrophin receptor activation. Cell. Mol. Neurobiol. 2007, 27, 585–594. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, X.; Li, S.; Chi, X.; Luo, A.; Zhao, Y. NR2B receptor- and calpain-mediated KCC2 cleavage resulted in cognitive deficiency exposure to isoflurane. Neurotoxicology 2020, 76, 75–83. [Google Scholar] [CrossRef]
- Go, J.; Park, T.S.; Han, G.H.; Park, H.Y.; Ryu, Y.K.; Kim, Y.H.; Hwang, J.H.; Choi, D.H.; Noh, J.R.; Hwang, D.Y.; et al. Piperlongumine decreases cognitive impairment and improves hippocampal function in aged mice. Int. J. Mol. Med. 2018, 42, 1875–1884. [Google Scholar] [CrossRef]
- Tu, F.P.; Li, J.X.; Li, Q.; Wang, J. Effects of hydrogen sulfide on cognitive dysfunction and NR2B in rats. J. Surg. Res. 2016, 205, 426–431. [Google Scholar] [CrossRef]
- Baudry, M.; Bi, X. Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 2016, 39, 235–245. [Google Scholar] [CrossRef]
- Blaesse, P.; Schmidt, T. K-Cl cotransporter KCC2—A moonlighting protein in excitatory and inhibitory synapse development and function. Pflug. Arch. Eur. J. Physiol. 2015, 467, 615–624. [Google Scholar] [CrossRef]
- Wang, M.; Xie, Y.; Qin, D. Proteolytic cleavage of proBDNF to mBDNF in neuropsychiatric and neurodegenerative diseases. Brain Res. Bull. 2021, 166, 172–184. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, J.; Min, S.; Ren, L.; Qin, P. Propofol alleviates electroconvulsive shock-induced memory impairment by modulating proBDNF/mBDNF ratio in depressive rats. Brain Res. 2016, 1642, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Fleitas, C.; Piñol-Ripoll, G.; Marfull, P.; Rocandio, D.; Ferrer, I.; Rampon, C.; Egea, J.; Espinet, C. proBDNF is modified by advanced glycation end products in Alzheimer’s disease and causes neuronal apoptosis by inducing p75 neurotrophin receptor processing. Mol. Brain 2018, 11, 68. [Google Scholar] [CrossRef]
- Gelle, T.; Samey, R.A.; Plansont, B.; Bessette, B.; Jauberteau-Marchan, M.O.; Lalloué, F.; Girard, M. BDNF and pro-BDNF in serum and exosomes in major depression: Evolution after antidepressant treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 109, 110229. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, S.; Li, C.; Lu, N.; Yue, Y.; Yin, Y.; Zhang, Y.; Zhi, X.; Zhang, D.; Yuan, Y. The serum protein levels of the tPA-BDNF pathway are implicated in depression and antidepressant treatment. Transl. Psychiatry 2017, 7, e1079. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, C.; Chen, J.; Su, Y.; Zhou, R.; Wang, F.; Xia, W.; Huang, J.; Wang, Z.; Hu, Y.; et al. Ratio of mBDNF to proBDNF for Differential Diagnosis of Major Depressive Disorder and Bipolar Depression. Mol. Neurobiol. 2017, 54, 5573–5582. [Google Scholar] [CrossRef]
- Bai, Y.Y.; Ruan, C.S.; Yang, C.R.; Li, J.Y.; Kang, Z.L.; Zhou, L.; Liu, D.; Zeng, Y.Q.; Wang, T.H.; Tian, C.F.; et al. ProBDNF Signaling Regulates Depression-Like Behaviors in Rodents under Chronic Stress. Neuropsychopharmacology 2016, 41, 2882–2892. [Google Scholar] [CrossRef]
- Yang, C.R.; Zhang, X.Y.; Liu, Y.; Du, J.Y.; Liang, R.; Yu, M.; Zhang, F.Q.; Mu, X.F.; Li, F.; Zhou, L.; et al. Antidepressant Drugs Correct the Imbalance Between proBDNF/p75NTR/Sortilin and Mature BDNF/TrkB in the Brain of Mice with Chronic Stress. Neurotox. Res. 2020, 37, 171–182. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Ma, N.; Yan, D.; Wang, Y.; Zhao, X.; Zhang, Y.; Zhang, C. Effects of corticosterone on the expression of mature brain-derived neurotrophic factor (mBDNF) and proBDNF in the hippocampal dentate gyrus. Behav. Brain Res. 2019, 365, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.Y.; Kelliny, S.; Liu, L.C.; Al-Hawwas, M.; Zhou, X.F.; Bobrovskaya, L. Peripheral ProBDNF Delivered by an AAV Vector to the Muscle Triggers Depression-Like Behaviours in Mice. Neurotox. Res. 2020, 38, 626–639. [Google Scholar] [CrossRef]
- Qiao, H.; An, S.C.; Xu, C.; Ma, X.M. Role of proBDNF and BDNF in dendritic spine plasticity and depressive-like behaviors induced by an animal model of depression. Brain Res. 2017, 1663, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Liu, J.; Manaph, N.P.A.; Bobrovskaya, L.; Zhou, X.F. ProBDNF inhibits proliferation, migration and differentiation of mouse neural stem cells. Brain Res. 2017, 1668, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.S.; Raheem, S.A.; Alshawi, S.A.; Regan, J.M.; Feng, L.R.; Saligan, L.N. Induction of fatigue-like behavior by pelvic irradiation of male mice alters cognitive behaviors and BDNF expression. PLoS ONE 2020, 15, e0235566. [Google Scholar] [CrossRef] [PubMed]
- Senra, H.; Balaskas, K.; Mahmoodi, N.; Aslam, T. Experience of Anti-VEGF Treatment and Clinical Levels of Depression and Anxiety in Patients With Wet Age-Related Macular Degeneration. Am. J. Ophthalmol. 2017, 177, 213–224. [Google Scholar] [CrossRef]
- van der Aa, H.P.; van Rens, G.H.; Verbraak, F.D.; Bosscha, M.; Comijs, H.C.; van Nispen, R.M. Anxiety and depression in patients who receive anti-VEGF treatment and the usability and feasibility of e-mental health support: The E-PsEYE pilot study. Ophthalmic Physiol. Opt. J. Br. Coll. Ophthalmic Opt. 2021, 41, 808–819. [Google Scholar] [CrossRef]
- Sideromenos, S.; Lindtner, C.; Zambon, A.; Horvath, O.; Berger, A.; Pollak, D.D. VEGF Treatment Ameliorates Depression-Like Behavior in Adult Offspring After Maternal Immune Activation. Cells 2020, 9, 1048. [Google Scholar] [CrossRef]
- Deyama, S.; Duman, R.S. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 2020, 188, 172837. [Google Scholar] [CrossRef]
- Ryan, K.M.; McLoughlin, D.M. Vascular endothelial growth factor plasma levels in depression and following electroconvulsive therapy. Eur. Arch. Psychiatry Clin. Neurosci. 2018, 268, 839–848. [Google Scholar] [CrossRef]
- Valiuliene, G.; Valiulis, V.; Dapsys, K.; Vitkeviciene, A.; Gerulskis, G.; Navakauskiene, R.; Germanavicius, A. Brain stimulation effects on serum BDNF, VEGF, and TNFα in treatment-resistant psychiatric disorders. Eur. J. Neurosci. 2021, 53, 3791–3802. [Google Scholar] [CrossRef]
- Kranaster, L.; Blennow, K.; Zetterberg, H.; Sartorius, A. Reduced vascular endothelial growth factor levels in the cerebrospinal fluid in patients with treatment resistant major depression and the effects of electroconvulsive therapy—A pilot study. J. Affect. Disord. 2019, 253, 449–453. [Google Scholar] [CrossRef]
- Çakici, N.; Sutterland, A.L.; Penninx, B.; Dalm, V.A.; de Haan, L.; van Beveren, N.J.M. Altered peripheral blood compounds in drug-naïve first-episode patients with either schizophrenia or major depressive disorder: A meta-analysis. Brain Behav. Immun. 2020, 88, 547–558. [Google Scholar] [CrossRef]
- Pu, J.; Liu, Y.; Gui, S.; Tian, L.; Xu, S.; Song, X.; Zhong, X.; Chen, Y.; Chen, X.; Yu, Y.; et al. Vascular endothelial growth factor in major depressive disorder, schizophrenia, and bipolar disorder: A network meta-analysis. Psychiatry Res. 2020, 292, 113319. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.-T.; Cheng, Y.-S.; Chen, Y.-W.; Wu, C.-K.; Lin, P.-Y. Increased levels of vascular endothelial growth factor in patients with major depressive disorder: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.F.R.; Cohen, A.; Edberg, D.; Hoppensteadt, D.; Fareed, J.; Martin, B.; Halaris, A. Vascular endothelial growth factor in bipolar depression: A potential biomarker for diagnosis and treatment outcome prediction. Psychiatry Res. 2020, 284, 112781. [Google Scholar] [CrossRef]
- Xiao, W.; Zhan, Q.; Ye, F.; Tang, X.; Li, J.; Dong, H.; Sha, W.; Zhang, X. Elevated serum vascular endothelial growth factor in treatment-resistant schizophrenia treated with electroconvulsive therapy: Positive association with therapeutic effects. World J. Biol. Psychiatry 2019, 20, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Electroconvulsive therapy increases temporarily plasma vascular endothelial growth factor in patients with major depressive disorder. Brain Behav. 2021, 11, e02001. [Google Scholar] [CrossRef]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef]
- Luck, R.; Urban, S.; Karakatsani, A.; Harde, E.; Sambandan, S.; Nicholson, L.; Haverkamp, S.; Mann, R.; Martin-Villalba, A.; Schuman, E.M.; et al. VEGF/VEGFR2 signaling regulates hippocampal axon branching during development. Elife 2019, 8, e49818. [Google Scholar] [CrossRef]
- Han, W.; Song, X.; He, R.; Li, T.; Cheng, L.; Xie, L.; Chen, H.; Jiang, L. VEGF regulates hippocampal neurogenesis and reverses cognitive deficits in immature rats after status epilepticus through the VEGF R2 signaling pathway. Epilepsy Behav. 2017, 68, 159–167. [Google Scholar] [CrossRef]
- Kolshus, E.; Ryan, K.M.; Blackshields, G.; Smyth, P.; Sheils, O.; McLoughlin, D.M. Peripheral blood microRNA and VEGFA mRNA changes following electroconvulsive therapy: Implications for psychotic depression. Acta Psychiatr. Scand. 2017, 136, 594–606. [Google Scholar] [CrossRef]
- Van Den Bossche, M.J.A.; Emsell, L.; Dols, A.; Vansteelandt, K.; De Winter, F.L.; Van den Stock, J.; Sienaert, P.; Stek, M.L.; Bouckaert, F.; Vandenbulcke, M. Hippocampal volume change following ECT is mediated by rs699947 in the promotor region of VEGF. Transl. Psychiatry 2019, 9, 191. [Google Scholar] [CrossRef]
- Repple, J.; Meinert, S.; Bollettini, I.; Grotegerd, D.; Redlich, R.; Zaremba, D.; Burger, C.; Forster, K.; Dohm, K.; Stahl, F.; et al. Influence of electroconvulsive therapy on white matter structure in a diffusion tensor imaging study. Psychol. Med. 2020, 50, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Alba-Ferrara, L.; De Erausquin, G. What does anisotropy measure? Insights from increased and decreased anisotropy in selective fiber tracts in schizophrenia. Front. Integr. Neurosci. 2013, 7, 9. [Google Scholar] [CrossRef]
- Meinert, S.; Repple, J.; Nenadic, I.; Krug, A.; Jansen, A.; Grotegerd, D.; Förster, K.; Enneking, V.; Dohm, K.; Schmitt, S.; et al. Reduced fractional anisotropy in depressed patients due to childhood maltreatment rather than diagnosis. Neuropsychopharmacology 2019, 44, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.E.; Noble, J.W.; Krassioukov, A.; Boyd, L.A.; Eng, J.J. Decreased white matter fractional anisotropy is associated with poorer functional motor skills following spinal cord injury: A pilot study. Spinal. Cord. 2019, 57, 206–213. [Google Scholar] [CrossRef]
- Gryglewski, G.; Seiger, R.; Baldinger-Melich, P.; Unterholzner, J.; Spurny, B.; Vanicek, T.; Hahn, A.; Kasper, S.; Frey, R.; Lanzenberger, R. Changes in White Matter Microstructure After Electroconvulsive Therapy for Treatment-Resistant Depression. Int. J. Neuropsychopharmacol. 2020, 23, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Njau, S.; Joshi, S.H.; Espinoza, R.; Leaver, A.M.; Vasavada, M.; Marquina, A.; Woods, R.P.; Narr, K.L. Neurochemical correlates of rapid treatment response to electroconvulsive therapy in patients with major depression. J. Psychiatry Neurosci. 2017, 42, 6–16. [Google Scholar] [CrossRef]
- Lefebvre, D.; Langevin, L.M.; Jaworska, N.; Harris, A.D.; Lebel, R.M.; Jasaui, Y.; Kirton, A.; Wilkes, T.C.; Sembo, M.; Swansburg, R.; et al. A pilot study of hippocampal N-acetyl-aspartate in youth with treatment resistant major depression. J. Affect. Disord. 2017, 207, 110–113. [Google Scholar] [CrossRef]
- Smith, G.S.; Oeltzschner, G.; Gould, N.F.; Leoutsakos, J.S.; Nassery, N.; Joo, J.H.; Kraut, M.A.; Edden, R.A.E.; Barker, P.B.; Wijtenburg, S.A.; et al. Neurotransmitters and Neurometabolites in Late-Life Depression: A Preliminary Magnetic Resonance Spectroscopy Study at 7T. J. Affect. Disord. 2021, 279, 417–425. [Google Scholar] [CrossRef]
- Knudsen, M.K.; Near, J.; Blicher, A.B.; Videbech, P.; Blicher, J.U. Magnetic resonance (MR) spectroscopic measurement of γ-aminobutyric acid (GABA) in major depression before and after electroconvulsive therapy. Acta Neuropsychiatr. 2019, 31, 17–26. [Google Scholar] [CrossRef]
- Erchinger, V.J.; Ersland, L.; Aukland, S.M.; Abbott, C.C.; Oltedal, L. Magnetic Resonance Spectroscopy in Depressed Subjects Treated With Electroconvulsive Therapy-A Systematic Review of Literature. Front. Psychiatry 2021, 12, 608857. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Lai, S.; Yue, J.; Wang, Y.; Shan, Y.; Liao, X.; Chen, J.; Li, Z.; Chen, G.; Chen, F.; et al. The characteristic of cognitive impairments in patients with bipolar II depression and its association with N-acetyl aspartate of the prefrontal white matter. Ann. Transl. Med. 2020, 8, 1457. [Google Scholar] [CrossRef] [PubMed]
- Evjenth Sørhaug, O.J.; Brekke, N.; Renate Grüner, E.; Ersland, L.; Kessler, U.; Oedegaard, K.J.; Oltedal, L. 256. Hippocampal Tissue Properties, as Evaluated by Flair and Susceptibility Weighted Imaging in a Preliminary Sample of Patients Treated with ECT. Biol. Psychiatry 2017, 81, S105–S106. [Google Scholar] [CrossRef]
- Nordanskog, P.; Dahlstrand, U.; Larsson, M.R.; Larsson, E.-M.; Knutsson, L.; Johanson, A. Increase in Hippocampal Volume After Electroconvulsive Therapy in Patients With Depression: A Volumetric Magnetic Resonance Imaging Study. J. ECT 2010, 26, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Nuninga, J.O.; Mandl, R.C.W.; Froeling, M.; Siero, J.C.W.; Somers, M.; Boks, M.P.; Nieuwdorp, W.; Heringa, S.; Sommer, I.E.C. Vasogenic edema versus neuroplasticity as neural correlates of hippocampal volume increase following electroconvulsive therapy. Brain Stimul. 2020, 13, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Gyger, L.; Ramponi, C.; Mall, J.F.; Swierkosz-Lenart, K.; Stoyanov, D.; Lutti, A.; von Gunten, A.; Kherif, F.; Draganski, B. Temporal trajectory of brain tissue property changes induced by electroconvulsive therapy. NeuroImage 2021, 232, 117895. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Maruyama, S.; Boku, S.; Okazaki, S.; Kikuyama, H.; Mizoguchi, Y.; Monji, A.; Otsuka, I.; Sora, I.; Kanazawa, T.; Hishimoto, A.; et al. ATP and repetitive electric stimulation increases leukemia inhibitory factor expression in astrocytes: A potential role for astrocytes in the action mechanism of electroconvulsive therapy. Psychiatry Clin. Neurosci. 2020, 74, 311–317. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef]
- Ito, M.; Bolati, K.; Kinjo, T.; Ichimura, K.; Furuta, A.; McLoughlin, D.M.; Suzuki, T.; Arai, H. Electroconvulsive stimulation transiently enhances the permeability of the rat blood-brain barrier and induces astrocytic changes. Brain Res. Bull. 2017, 128, 92–97. [Google Scholar] [CrossRef]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef]
- Azis, I.A.; Hashioka, S.; Tsuchie, K.; Miyaoka, T.; Abdullah, R.A.; Limoa, E.; Arauchi, R.; Inoue, K.; Miura, S.; Izuhara, M.; et al. Electroconvulsive shock restores the decreased coverage of brain blood vessels by astrocytic endfeet and ameliorates depressive-like behavior. J. Affect. Disord. 2019, 257, 331–339. [Google Scholar] [CrossRef]
- Limoa, E.; Hashioka, S.; Miyaoka, T.; Tsuchie, K.; Arauchi, R.; Azis, I.A.; Wake, R.; Hayashida, M.; Araki, T.; Furuya, M.; et al. Electroconvulsive shock attenuated microgliosis and astrogliosis in the hippocampus and ameliorated schizophrenia-like behavior of Gunn rat. J. Neuroinflamm. 2016, 13, 230. [Google Scholar] [CrossRef] [PubMed]
- Giacobbe, J.; Pariante, C.M.; Borsini, A. The innate immune system and neurogenesis as modulating mechanisms of electroconvulsive therapy in pre-clinical studies. J. Psychopharmacol. 2020, 34, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, W.; Lu, Y.; Lu, Y.; Xu, L.; Fang, Q.; Wu, M.; Jia, M.; Wang, Y.; Dong, L.; et al. Aquaporin 4-Mediated Glutamate-Induced Astrocyte Swelling Is Partially Mediated through Metabotropic Glutamate Receptor 5 Activation. Front. Cell. Neurosci. 2017, 11, 116. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, D.; Jiang, D.; Kulikowicz, E.; Tekes, A.; Liu, P.; Qin, Q.; Koehler, R.C.; Aggarwal, M.; Zhang, J.; et al. Fractional anisotropy from diffusion tensor imaging correlates with acute astrocyte and myelin swelling in neonatal swine models of excitotoxic and hypoxic-ischemic brain injury. J. Comp. Neurol. 2021, 529, 2750–2770. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Zielińska, M. Mechanisms of Excessive Extracellular Glutamate Accumulation in Temporal Lobe Epilepsy. Neurochem. Res. 2017, 42, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Takamiya, A.; Noda, Y.; Horita, N.; Wada, M.; Tsugawa, S.; Plitman, E.; Sano, Y.; Tarumi, R.; ElSalhy, M.; et al. Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol. Psychiatry 2019, 24, 952–964. [Google Scholar] [CrossRef]
- Xu, P.; Chen, A.; Li, Y.; Xing, X.; Lu, H. Medial prefrontal cortex in neurological diseases. Physiol. Genom. 2019, 51, 432–442. [Google Scholar] [CrossRef]
- Jing, Y.; Zhao, N.; Deng, X.P.; Feng, Z.J.; Huang, G.F.; Meng, M.; Zang, Y.F.; Wang, J. Pregenual or subgenual anterior cingulate cortex as potential effective region for brain stimulation of depression. Brain Behav. 2020, 10, e01591. [Google Scholar] [CrossRef]
- Ermis, C.; Aydin, B.; Kucukguclu, S.; Yurt, A.; Renshaw, P.F.; Yildiz, A. Association Between Anterior Cingulate Cortex Neurochemical Profile and Clinical Remission After Electroconvulsive Treatment in Major Depressive Disorder: A Longitudinal 1H Magnetic Resonance Spectroscopy Study. J. ECT 2021, 37, 263–269. [Google Scholar] [CrossRef]
- Anderson, I.M.; Blamire, A.; Branton, T.; Clark, R.; Downey, D.; Dunn, G.; Easton, A.; Elliott, R.; Elwell, C.; Hayden, K.; et al. Ketamine augmentation of electroconvulsive therapy to improve neuropsychological and clinical outcomes in depression (Ketamine-ECT): A multicentre, double-blind, randomised, parallel-group, superiority trial. Lancet Psychiatry 2017, 4, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, A.; Rüst, C.A.; Riegger, H.; Gysi, B.; Tran, F.; Brügger, J.; Huber, J.; Toft, K.; Surbeck, M.; Schmid, H.R.; et al. Ketamine vs. haloperidol for prevention of cognitive dysfunction and postoperative delirium: A phase IV multicentre randomised placebo-controlled double-blind clinical trial. J. Clin. Anesth. 2021, 68, 110099. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Piguel, N.H.; Khalatyan, N.; Dionisio, L.E.; Savas, J.N.; Penzes, P. Homer1 promotes dendritic spine growth through ankyrin-G and its loss reshapes the synaptic proteome. Mol. Psychiatry 2021, 26, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Ismail, Z.; Hatta, N.H.; Baharuddin, N.; Hapidin, H.; Get Bee, Y.T.; Yap, E.; Pakri Mohamed, R.M. Alcohol Addiction- Metabotropic Glutamate Receptor Subtype 5 and its Ligands: How They All Come Together? Curr. Drug Targets 2018, 19, 907–915. [Google Scholar] [CrossRef]
- Kaastrup Müller, H.; Orlowski, D.; Reidies Bjarkam, C.; Wegener, G.; Elfving, B. Potential roles for Homer1 and Spinophilin in the preventive effect of electroconvulsive seizures on stress-induced CA3c dendritic retraction in the hippocampus. Eur. Neuropsychopharmacol. 2015, 25, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Holz, A.; Mülsch, F.; Schwarz, M.K.; Hollmann, M.; Döbrössy, M.D.; Coenen, V.A.; Bartos, M.; Normann, C.; Biber, K.; van Calker, D.; et al. Enhanced mGlu5 Signaling in Excitatory Neurons Promotes Rapid Antidepressant Effects via AMPA Receptor Activation. Neuron 2019, 104, 338–352.e337. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ginet, V.; Lopatar, J.; Montana, V.; Pucci, L.; Spagnuolo, P.; Zehnder, T.; Grubišic, V.; Truttman, A.; Sala, C.; et al. Homer1 Scaffold Proteins Govern Ca2+ Dynamics in Normal and Reactive Astrocytes. Cerebral. Cortex. 2017, 27, 2365–2384. [Google Scholar] [CrossRef]
- Kumar, J.; Hapidin, H.; Bee, Y.-T.G.; Ismail, Z. Effects of the mGluR5 antagonist MPEP on ethanol withdrawal induced anxiety-like syndrome in rats. Behav. Brain Funct. 2013, 9, 43. [Google Scholar] [CrossRef]
- Nardecchia, F.; Orlando, R.; Iacovelli, L.; Colamartino, M.; Fiori, E.; Leuzzi, V.; Piccinin, S.; Nistico, R.; Puglisi-Allegra, S.; Di Menna, L.; et al. Targeting mGlu5 Metabotropic Glutamate Receptors in the Treatment of Cognitive Dysfunction in a Mouse Model of Phenylketonuria. Front. Neurosci. 2018, 12, 154. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ECT Electrode Placement | Brain Changes | Cognitive Outcome | Reference |
|---|---|---|---|
| Bifrontal | Increased cortical thickness in the left inferior parietal gyrus and reduced surface area of right inferior temporal gyrus | Impaired delayed memory | [83] |
| Bitemporal | Increased volume in bilateral hippocampus | Slower processing speed and impaired divided attention ability, impaired delayed memory | [84] |
| Bitemporal | (Immediately after ECT series) increased volume in right and left DG | Reduced delayed memory performance | [17] |
| (At 6-month follow-up) reduced volume in right DG | Improved delayed memory performance | ||
| RUL | Larger absolute increase in right hippocampal volume | (Assessment at 6 months) less improvement in visual memory | [85] |
| Switched to BT after failed RUL | Larger absolute increase in right hippocampal volume | (Assessment at 6 months) less improvement in semantic memory and verbal memory, less improvement in global cognitive functioning | |
| Bifrontal | Reduced RSFC between left hippocampal middle (cognitive) subregion and bilateral angular gyrus | Impaired performance on the verbal fluency test | [21] |
| Bifrontal (for major depressive episode) and bitemporal (schizophrenia spectrum disorder) | Increased volume in the hippocampus and amygdala | Lower Repeatable Battery of the Assessment of Neuropsychological Status (RBANS) scoring | [86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad Hariza, A.M.; Mohd Yunus, M.H.; Murthy, J.K.; Wahab, S. Clinical Improvement in Depression and Cognitive Deficit Following Electroconvulsive Therapy. Diagnostics 2023, 13, 1585. https://doi.org/10.3390/diagnostics13091585
Ahmad Hariza AM, Mohd Yunus MH, Murthy JK, Wahab S. Clinical Improvement in Depression and Cognitive Deficit Following Electroconvulsive Therapy. Diagnostics. 2023; 13(9):1585. https://doi.org/10.3390/diagnostics13091585
Chicago/Turabian StyleAhmad Hariza, Ahmad Mus’ab, Mohd Heikal Mohd Yunus, Jaya Kumar Murthy, and Suzaily Wahab. 2023. "Clinical Improvement in Depression and Cognitive Deficit Following Electroconvulsive Therapy" Diagnostics 13, no. 9: 1585. https://doi.org/10.3390/diagnostics13091585
APA StyleAhmad Hariza, A. M., Mohd Yunus, M. H., Murthy, J. K., & Wahab, S. (2023). Clinical Improvement in Depression and Cognitive Deficit Following Electroconvulsive Therapy. Diagnostics, 13(9), 1585. https://doi.org/10.3390/diagnostics13091585