

Average Nucleotide Identity and Digital DNA-DNA Hybridization Analysis Following PromethION Nanopore-Based Whole Genome Sequencing Allows for Accurate Prokaryotic Typing

 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. Culture Conditions
2.3. Antibiotic Susceptibility Testing
2.4. DNA Extraction
2.5. Assessment of DNA Fragmentation and Concentration
2.6. WGS with Oxford Nanopore Technologies PromethION
2.7. Construction of a New Bioinformatics Pipeline
2.8. Genome Quality Statistics for the WGS Assemblies
2.9. WGS-Based Typing of Bacterial Genomes
2.10. Genomic Screening for Antibiotic Resistance Markers and Comparison with Phenotype
3. Results
3.1. Characterization of the DNA Extracts
3.2. Protocol Optimization and Library Sequencing Features
3.3. Characterization of the WGS Data
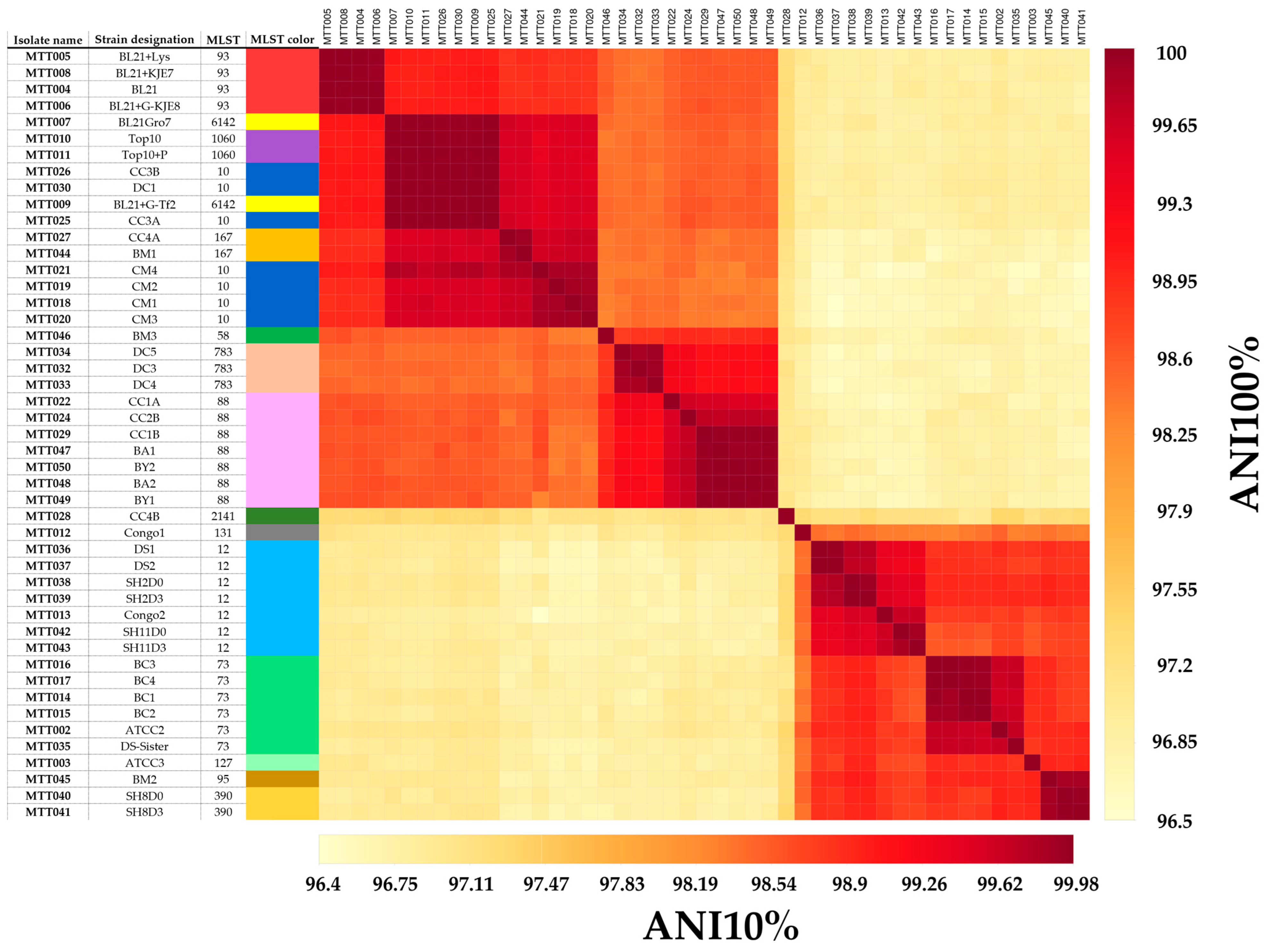
3.3.1. ANI and DDH Percentage Identities Compared to MLST
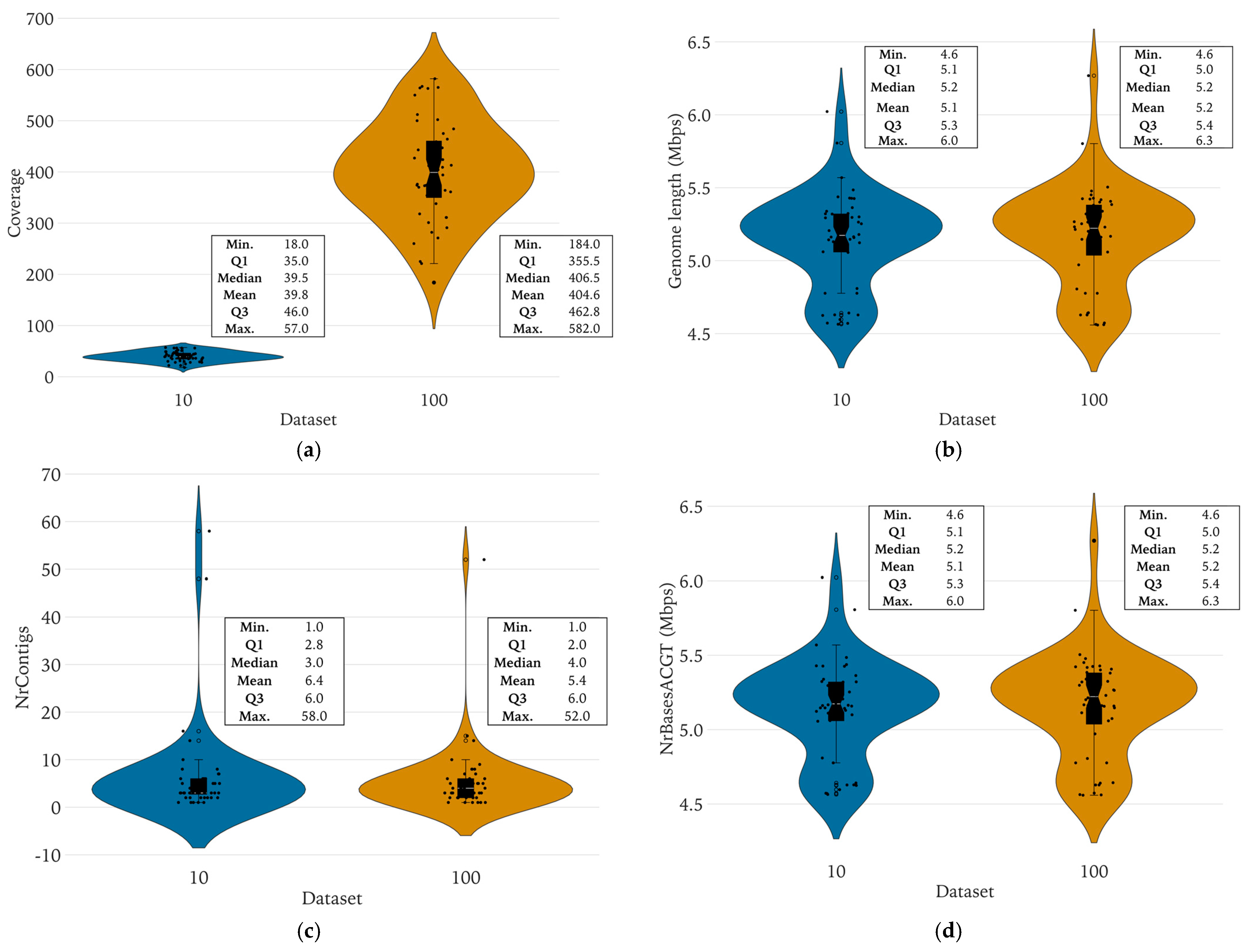
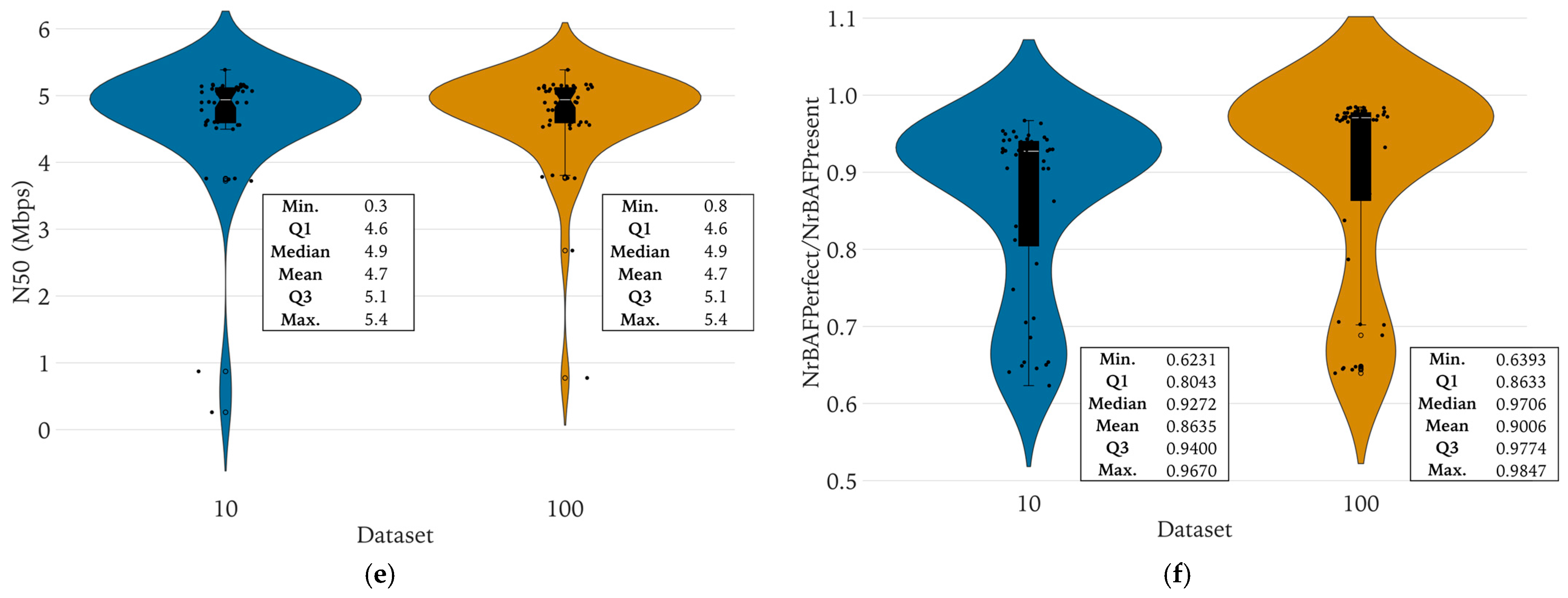
3.3.2. Determination of the Minimal Sequencing Coverage Required for Qualitative Analyses
3.3.3. Pairwise Analysis of WGS Features between the Complete and the Reduced Datasets
3.3.4. Pairwise Assessment of Strain Typing with MLST and ANI for the Complete and the Reduced Datasets
3.3.5. Pairwise Analysis of Antibiotic Resistance Genes in the Complete and Reduced Datasets
4. Discussion
4.1. An Optimized Protocol for the PromethION Whole-Genome Sequencing of Prokaryotes
4.2. ANI and DDH Are Robust Strain Typing Methods for E. coli
4.3. WGS-Based Prediction of Antibiotic Resistance Compared to MIC Assay Predicts with >90% Accuracy for Some Classes of Antibiotics
4.4. A Minimal Sequencing Coverage of 12× Was Required to Maintain Key Features
4.5. There Were No Significant Differences in Genomic Key Features, Typing, and Antibiotic Resistance Prediction between the Complete and Reduced Datasets
4.6. The Choice of MLST Classification Tool Determines the Robustness of the Results
5. Conclusions
Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bertelli, C.; Greub, G. Rapid bacterial genome sequencing: Methods and applications in clinical microbiology. Clin. Microbiol. Infect. 2013, 19, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Portmann, A.C.; Fournier, C.; Gimonet, J.; Ngom-Bru, C.; Barretto, C.; Baert, L. A Validation Approach of an End-to-End Whole Genome Sequencing Workflow for Source Tracking of Listeria monocytogenes and Salmonella enterica. Front. Microbiol. 2018, 9, 446. [Google Scholar] [CrossRef]
- Oniciuc, E.A.; Likotrafiti, E.; Alvarez-Molina, A.; Prieto, M.; Santos, J.A.; Alvarez-Ordóñez, A. The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain. Genes 2018, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Petkau, A.; Mabon, P.; Sieffert, C.; Knox, N.C.; Cabral, J.; Iskander, M.; Iskander, M.; Weedmark, K.; Zaheer, R.; Katz, L.S.; et al. SNVPhyl: A single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microb. Genom. 2017, 3, e000116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Uelze, L.; Grützke, J.; Borowiak, M.; Hammerl, J.A.; Juraschek, K.; Deneke, C.; Tausch, S.H.; Malorny, B. Typing methods based on whole genome sequencing data. One Health Outlook 2020, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Maiden, M.C.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Dougherty, K.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.S.; Foster, J.T. Incongruence between multi-locus sequence analysis (MLSA) and whole-genome-based phylogenies: Pseudomonas syringae pathovar pisi as a cautionary tale. Mol. Plant Pathol. 2014, 15, 461–465. [Google Scholar] [CrossRef]
- Rodriguez, R.L.; Conrad, R.E.; Viver, T.; Feistel, D.J.; Lindner, B.G.; Venter, S.N.; Orellana, L.H.; Amann, R.; Rossello-Mora, R.; Konstantinidis, K.T. An ANI gap within bacterial species that advances the definitions of intra-species units. mBio 2024, 15, e0269623. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Reller, L.B.; Weinstein, M.; Jorgensen, J.H.; Ferraro, M.J. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar] [CrossRef]
- Bravo, L.T.; Procop, G.W. Recent advances in diagnostic microbiology. Semin. Hematol. 2009, 46, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Cools, P.; Haelters, J.; Lopes dos Santos Santiago, G.; Claeys, G.; Boelens, J.; Leroux-Roels, I.; Vaneechoutte, M.; Deschaght, P. Edwardsiella tarda sepsis in a live-stranded sperm whale (Physeter macrocephalus). Vet. Microbiol. 2013, 166, 311–315. [Google Scholar] [CrossRef] [PubMed]
- [x] ISO 20776-1; Susceptibility Testing of Infectious Agents and Evaluation of Performance of Antimicrobial Susceptibility Test Devices—Part 1: Broth Microdilution Reference Method for Testing the In Vitro Activity of Antimicrobial Agents against Rapidly Growing Aerobic Bacteria Involved in Infectious Diseases. International Organization for Standardization: Geneva, Switzerland, 2019.
- Matuschek, E.; Brown, D.F.; Kahlmeter, G. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol. Infect. 2014, 20, O255–O266. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Turner, D.; Mesirov, J.P. igv.js: An embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Serres, M.H.; Gopal, S.; Nahum, L.A.; Liang, P.; Gaasterland, T.; Riley, M. A functional update of the Escherichia coli K-12 genome. Genome Biol. 2001, 2, Research0035. [Google Scholar] [CrossRef]
- Bartual, S.G.; Seifert, H.; Hippler, C.; Luzon, M.A.; Wisplinghoff, H.; Rodríguez-Valera, F. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J. Clin. Microbiol. 2005, 43, 4382–4390. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Griffiths, D.; Fawley, W.; Kachrimanidou, M.; Bowden, R.; Crook, D.W.; Fung, R.; Golubchik, T.; Harding, R.M.; Jeffery, K.J.; Jolley, K.A.; et al. Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 2010, 48, 770–778. [Google Scholar] [CrossRef]
- Jaureguy, F.; Landraud, L.; Passet, V.; Diancourt, L.; Frapy, E.; Guigon, G.; Carbonnelle, E.; Lortholary, O.; Clermont, O.; Denamur, E.; et al. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genom. 2008, 9, 560. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Lemee, L.; Dhalluin, A.; Pestel-Caron, M.; Lemeland, J.F.; Pons, J.L. Multilocus sequence typing analysis of human and animal Clostridium difficile isolates of various toxigenic types. J. Clin. Microbiol. 2004, 42, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2021, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van. Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Humphries, R.M.; Ambler, J.; Mitchell, S.L.; Castanheira, M.; Dingle, T.; Hindler, J.A.; Koeth, L.; Sei, K.; Development, C.M.; Standardization Working Group of the Subcommittee on Antimicrobial Susceptibility, T. CLSI Methods Development and Standardization Working Group Best Practices for Evaluation of Antimicrobial Susceptibility Tests. J. Clin. Microbiol. 2018, 56, e01934-17. [Google Scholar] [CrossRef] [PubMed]
- Stortchevoi, A.; Kamelamela, N.; Levine, S.S. SPRI Beads-based Size Selection in the Range of 2–10kb. J. Biomol. Tech. 2020, 31, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Swerdlow, H.; Turner, D.J. Improved protocols for the illumina genome analyzer sequencing system. Curr. Protoc. Hum. Genet. 2009, 62, 18.2.1–18.2.27. [Google Scholar] [CrossRef]
- Borgström, E.; Lundin, S.; Lundeberg, J. Large scale library generation for high throughput sequencing. PLoS ONE 2011, 6, e19119. [Google Scholar] [CrossRef]
- Lundin, S.; Stranneheim, H.; Pettersson, E.; Klevebring, D.; Lundeberg, J. Increased throughput by parallelization of library preparation for massive sequencing. PLoS ONE 2010, 5, e10029. [Google Scholar] [CrossRef]
- DeAngelis, M.M.; Wang, D.G.; Hawkins, T.L. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 1995, 23, 4742–4743. [Google Scholar] [CrossRef]
- Pearce, M.E.; Alikhan, N.F.; Dallman, T.J.; Zhou, Z.; Grant, K.; Maiden, M.C.J. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 2018, 274, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vincent, C.; Usongo, V.; Berry, C.; Tremblay, D.M.; Moineau, S.; Yousfi, K.; Doualla-Bell, F.; Fournier, E.; Nadon, C.; Goodridge, L.; et al. Comparison of advanced whole genome sequence-based methods to distinguish strains of Salmonella enterica serovar Heidelberg involved in foodborne outbreaks in Québec. Food Microbiol. 2018, 73, 99–110. [Google Scholar] [CrossRef]
- Henri, C.; Leekitcharoenphon, P.; Carleton, H.A.; Radomski, N.; Kaas, R.S.; Mariet, J.F.; Felten, A.; Aarestrup, F.M.; Gerner Smidt, P.; Roussel, S.; et al. An Assessment of Different Genomic Approaches for Inferring Phylogeny of Listeria monocytogenes. Front. Microbiol. 2017, 8, 2351. [Google Scholar] [CrossRef] [PubMed]
- Labbé, G.; Kruczkiewicz, P.; Robertson, J.; Mabon, P.; Schonfeld, J.; Kein, D.; Rankin, M.A.; Gopez, M.; Hole, D.; Son, D.; et al. Rapid and accurate SNP genotyping of clonal bacterial pathogens with BioHansel. Microb. Genom. 2021, 7, e000651. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Griswold, T.; Williams-Newkirk, A.J.; Wagner, D.; Petkau, A.; Sieffert, C.; Van Domselaar, G.; Deng, X.; Carleton, H.A. A Comparative Analysis of the Lyve-SET Phylogenomics Pipeline for Genomic Epidemiology of Foodborne Pathogens. Front. Microbiol. 2017, 8, 375. [Google Scholar] [CrossRef]
- Bush, S.J.; Foster, D.; Eyre, D.W.; Clark, E.L.; De Maio, N.; Shaw, L.P.; Stoesser, N.; Peto, T.E.A.; Crook, D.W.; Walker, A.S. Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism-calling pipelines. GigaScience 2020, 9, giaa007. [Google Scholar] [CrossRef]
- Vanstokstraeten, R.; Piérard, D.; Crombé, F.; De Geyter, D.; Wybo, I.; Muyldermans, A.; Seyler, L.; Caljon, B.; Janssen, T.; Demuyser, T. Genotypic resistance determined by whole genome sequencing versus phenotypic resistance in 234 Escherichia coli isolates. Sci. Rep. 2023, 13, 449. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.H.; McDermott, P.F.; Li, C.; Chen, Y.; Tadesse, D.A.; Mukherjee, S.; Bodeis-Jones, S.; Kabera, C.; Gaines, S.A.; Loneragan, G.H.; et al. WGS accurately predicts antimicrobial resistance in Escherichia coli. J. Antimicrob. Chemother. 2015, 70, 2763–2769. [Google Scholar] [CrossRef]
- Van Honacker, E.; Vandendriessche, S.; Coorevits, L.; Verhasselt, B.; Boelens, J. Impact of the introduction of EUCAST’s concept of “area of technical uncertainty”. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2022, 41, 203–207. [Google Scholar] [CrossRef]
- van der Putten, B.C.L.; Remondini, D.; Pasquini, G.; Janes, V.A.; Matamoros, S.; Schultsz, C. Quantifying the contribution of four resistance mechanisms to ciprofloxacin MIC in Escherichia coli: A systematic review. J. Antimicrob. Chemother. 2019, 74, 298–310. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Alikhan, N.-F.; Carleton, H.A.; Seemann, T.; Keane, J.A.; Katz, L.S. Comparison of classical multi-locus sequence typing software for next-generation sequencing data. Microb. Genom. 2017, 3, e000124. [Google Scholar] [CrossRef] [PubMed]
- Rode, C.K.; Melkerson-Watson, L.J.; Johnson, A.T.; Bloch, C.A. Type-specific contributions to chromosome size differences in Escherichia coli. Infect. Immun. 1999, 67, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore Technologies. Native Barcoding Genomic DNA (with EXP-NBD196 and SQK-LSK109); Oxford Nanopore Technologies: Oxford, UK, 2021; p. 27. [Google Scholar]
- Oxford Nanopore Technologies. Lambda Control Experiment (SQK-LSK109); Oxford Nanopore Technologies: Oxford, UK, 2019; p. 23. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| MLST | Ngenomes | %ANImin | %DDHmin | Remark |
|---|---|---|---|---|
| ST93 | 4 | 99.94 | 100 | Aligns with a single ANI/DDH cluster |
| ST6142 | 2 | 99.95 | 99.90 | Aligns with an ANI/DDH cluster also containing ST1060, ST10 |
| ST1060 | 2 | 99.98 | 100 | Aligns with an ANI/DDH cluster also containing ST6142 and ST10. |
| ST10 | 6 | 99.87 | 98.9 | Aligns with two ANI/DDH clusters with one cluster containing only ST10 and one cluster containing ST6142 and ST1060. |
| ST167 | 2 | 99.91 | 99.70 | Aligns with a single ANI/DDH cluster and is closely related to ANI/DDH clusters containing ST6142, ST1060, and ST10. |
| ST88 | 8 | 99.36 | 95.00 | Aligns with a single ANI/DDH cluster. Further discrimination is possible based on ANI/DDH. |
| ST783 | 3 | 99.90 | 99.60 | Aligns with a single ANI/DDH cluster. |
| ST12 | 7 | 99.32 | 94.10 | Aligns with a single ANI/DDH cluster. Further discrimination is possible based on ANI/DDH. |
| ST73 | 6 | 99.62 | 96.70 | Aligns with a single ANI/DDH cluster. Further discrimination is possible based on ANI/DDH. |
| ST390 | 2 | 99.96 | 100 | Aligns with an ANI/DDH cluster also containing ST95 (N = 1). |
| % Susceptible Phenotype (MIC) | % Intermediate Resistant phenotype (MIC) | % Resistant Phenotype (MIC) | % Susceptible Genotype | % Resistant Genotype | % Categorical Agreement | Minor Errors (%) | Major Errors (%) | Very major Errors (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100% | 10% | 100% | 10% | 100% | 10% | 100% | 10% | 100% | 10% | 100% | 10% | ||||
| PENICILLINS | |||||||||||||||
| Amoxicillin | 31.3 | 0.0 | 68.8 | 33.3 | 31.3 | 66.7 | 68.8 | 97.9 | 100.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.1 | 0.0 |
| Amoxicillin-Clavulanic Acid | 45.8 | 0.0 | 54.2 | 77.1 | 77.1 | 22.9 | 22.9 | 68.8 | 68.8 | 0.0 | 0.0 | 0.0 | 0.0 | 31.3 | 31.3 |
| Piperacillin-Tazobactam | 75.0 | 0.0 | 25.0 | 100.0 | 97.9 | 0.0 | 2.1 | 75.0 | 77.1 | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 22.9 |
| CEPHALOSPORINS | |||||||||||||||
| Cefepime | 91.7 | 4.2 | 4.2 | 81.3 | 81.3 | 18.7 | 18.8 | 81.3 | 79.2 | 0.0 | 2.1 | 16.7 | 16.7 | 2.1 | 2.1 |
| Ceftazidime | 62.5 | 0.0 | 37.5 | 58.3 | 60.4 | 41.7 | 39.6 | 95.8 | 93.8 | 0.0 | 0.0 | 4.2 | 4.2 | 0.0 | 2.1 |
| CARBAPENEMS | |||||||||||||||
| Meropenem | 100.0 | 0.0 | 0.0 | 100.0 | 100.0 | 0.0 | 0.0 | 100.0 | 100.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| MONOBACTAMS | |||||||||||||||
| Aztreonam | 58.3 | 4.2 | 37.5 | 81.3 | 81.3 | 18.7 | 18.8 | 68.8 | 68.8 | 4.2 | 4.2 | 4.2 | 4.2 | 22.9 | 22.9 |
| FLUOROQUINOLONES | |||||||||||||||
| Ciprofloxacin | 72.9 | 4.2 | 22.9 | 75.0 | 72.9 | 25.0 | 27.1 | 79.2 | 81.3 | 2.1 | 2.1 | 10.4 | 10.4 | 8.3 | 6.3 |
| AMINOGLYCOSIDES | |||||||||||||||
| Amikacin | 91.7 | 0.0 | 8.3 | 91.7 | 89.6 | 8.3 | 10.4 | 95.8 | 97.9 | 0.0 | 0.0 | 2.1 | 2.1 | 2.1 | 0.0 |
| Tobramycin | 75.0 | 0.0 | 25.0 | 77.1 | 77.1 | 22.9 | 22.9 | 93.8 | 93.8 | 0.0 | 0.0 | 2.1 | 2.1 | 4.2 | 4.2 |
| TETRACYCLINES | |||||||||||||||
| Tigecycline | 93.8 | 0.0 | 6.3 | 100.0 | 100.0 | 0.0 | 0.0 | 93.8 | 93.8 | 0.0 | 0.0 | 0.0 | 0.0 | 6.3 | 6.3 |
| MISCELLANEOUS AGENTS | |||||||||||||||
| Colistin | 93.8 | 0.0 | 6.3 | 100.0 | 100.0 | 0.0 | 0.0 | 93.8 | 93.8 | 0.0 | 0.0 | 0.0 | 0.0 | 6.3 | 6.3 |
| Trimethoprim-Sulfamethoxazole | 70.8 | 0.0 | 29.2 | 72.9 | 70.8 | 27.1 | 29.2 | 97.9 | 100.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.1 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Versmessen, N.; Mispelaere, M.; Vandekerckhove, M.; Hermans, C.; Boelens, J.; Vranckx, K.; Van Nieuwerburgh, F.; Vaneechoutte, M.; Hulpiau, P.; Cools, P. Average Nucleotide Identity and Digital DNA-DNA Hybridization Analysis Following PromethION Nanopore-Based Whole Genome Sequencing Allows for Accurate Prokaryotic Typing. Diagnostics 2024, 14, 1800. https://doi.org/10.3390/diagnostics14161800
Versmessen N, Mispelaere M, Vandekerckhove M, Hermans C, Boelens J, Vranckx K, Van Nieuwerburgh F, Vaneechoutte M, Hulpiau P, Cools P. Average Nucleotide Identity and Digital DNA-DNA Hybridization Analysis Following PromethION Nanopore-Based Whole Genome Sequencing Allows for Accurate Prokaryotic Typing. Diagnostics. 2024; 14(16):1800. https://doi.org/10.3390/diagnostics14161800
Chicago/Turabian StyleVersmessen, Nick, Marieke Mispelaere, Marjolein Vandekerckhove, Cedric Hermans, Jerina Boelens, Katleen Vranckx, Filip Van Nieuwerburgh, Mario Vaneechoutte, Paco Hulpiau, and Piet Cools. 2024. "Average Nucleotide Identity and Digital DNA-DNA Hybridization Analysis Following PromethION Nanopore-Based Whole Genome Sequencing Allows for Accurate Prokaryotic Typing" Diagnostics 14, no. 16: 1800. https://doi.org/10.3390/diagnostics14161800
APA StyleVersmessen, N., Mispelaere, M., Vandekerckhove, M., Hermans, C., Boelens, J., Vranckx, K., Van Nieuwerburgh, F., Vaneechoutte, M., Hulpiau, P., & Cools, P. (2024). Average Nucleotide Identity and Digital DNA-DNA Hybridization Analysis Following PromethION Nanopore-Based Whole Genome Sequencing Allows for Accurate Prokaryotic Typing. Diagnostics, 14(16), 1800. https://doi.org/10.3390/diagnostics14161800