

The Impact of Liquid Biopsy in Advanced Ovarian Cancer Care

 , , , , , , , ,
, , , , , , , ,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Patient Characteristics
2.2. Genomic DNA and cfDNA Isolation
2.3. Sequencing
2.4. Bioinformatic Analysis for Mutation Detection and Evaluation
2.5. Primer and Probe Design for ddPCR
- dHsaMDV2010105 for the TP53R175H mutation;
- dHsaMDV2010127 for the TP53R248Q mutation;
- dHsaMDV2510542 for the TP53G245D mutation;
- dHsaMDS99673883 for the BRCA1 E272*(stop) mutation;
- dHsaMDS544377207 for the TP53c.994C-1G mutation.
2.6. Droplet Digital PCR (ddPCR)
2.7. Statistical Analysis
3. Results
3.1. Overview
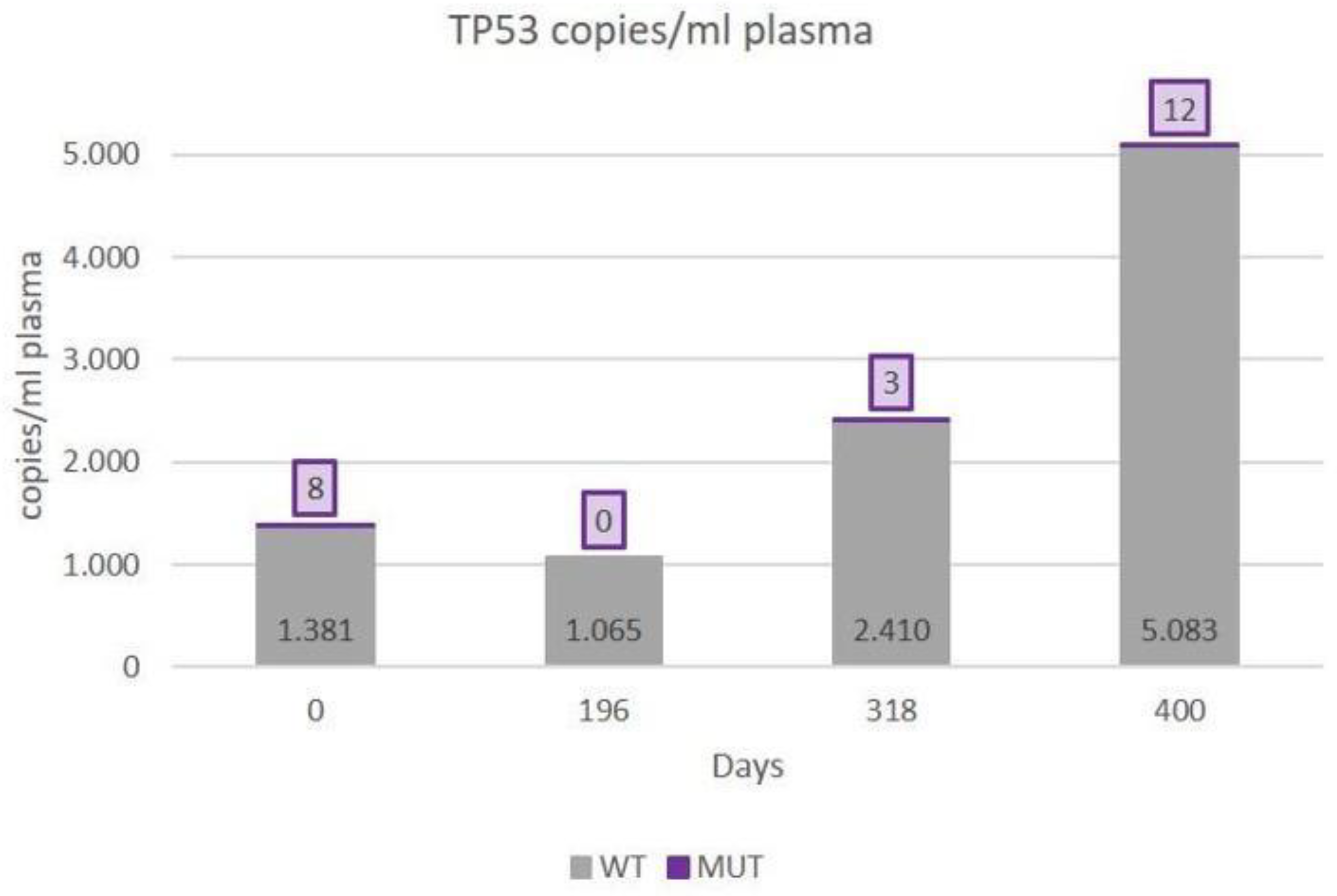
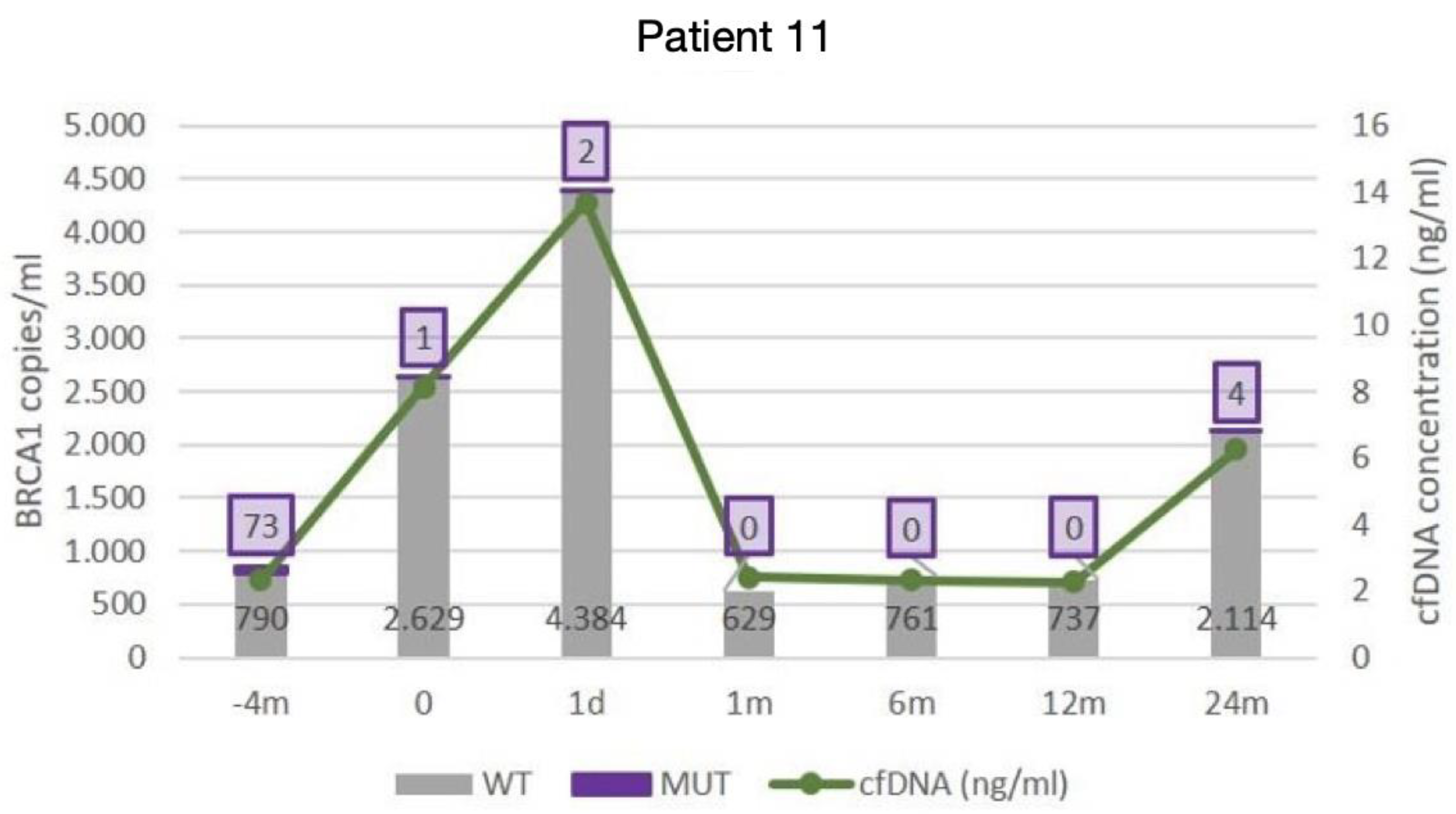
3.2. ctDNA Mutations and the Clinical Outcome
- (A)
- First patient with complete records. (Patient with clear disease progression on treatment)
- (B)
- Second patient with complete records. (Patient with clear disease progression on treatment)
- (C)
- Third patient with complete records. (Patient with stable disease monitoring)
- (D)
- Fourth patient with complete records. (Patient with evidence of relapse only in cfDNA, indicating changes in tumour cell subpopulations)
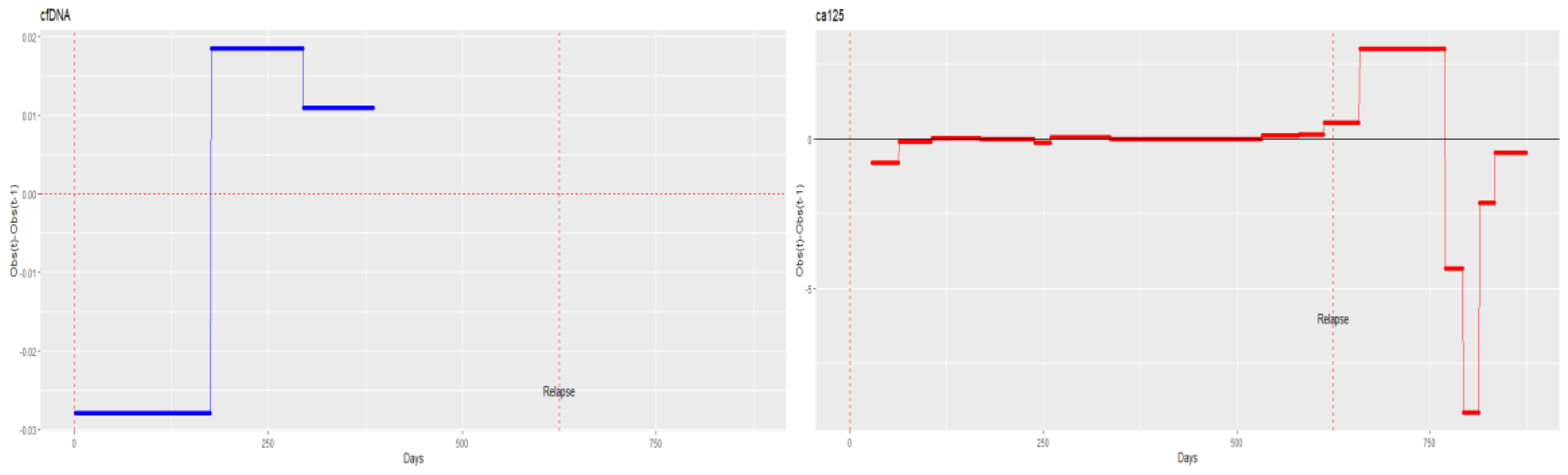
3.3. Correlations between Individualised ctDNA Mutations and Serum Biomarkers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winter, W.E., III; Maxwell, G.L.; Tian, C.; Carlson, J.W.; Ozols, R.F.; Rose, P.G.; Markman, M.; Armstrong, D.K.; Muggia, F.; McGuire, W.P. Prognostic factors for stage III epithelial ovarian cancer: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2007, 25, 3621–3627. [Google Scholar] [CrossRef]
- Llueca, A.; Serra, A.; Maiocchi, K.; Delgado, K.; Jativa, R.; Gomez, L.; Escrig, J. Predictive model for major complications after extensive abdominal surgery in primary advanced ovarian cancer. Int. J. Womens Health. 2019, 11, 161–167. [Google Scholar] [CrossRef]
- Llueca, A.; Serra, A.; Climent, M.T.; Segarra, B.; Maazouzi, Y.; Soriano, M.; Escrig, J.; on behalf MUAPOS Working Group. Outcome quality standards in advanced ovarian cancer surgery. World J. Surg. Oncol. 2020, 18, 309. [Google Scholar] [CrossRef]
- Garzon, S.; Laganà, A.S.; Casarin, J.; Raffaelli, R.; Cromi, A.; Franchi, M.; Barra, F.; Alkatout, I.; Ferrero, S.; Ghezzi, F. Secondary and tertiary ovarian cancer recurrence: What is the best management? Gland. Surg. 2020, 9, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.T.; Serra, A.; Llueca, M.; Llueca, A. Surgery in Recurrent Ovarian Cancer: A Meta-Analysis. Cancers 2023, 15, 3470. [Google Scholar] [CrossRef]
- Van Der Burg, M.E.L.; Lammes, F.B.; Verweij, J. The role of CA 125 in the early diagnosis of progressive disease in ovarian cancer. Ann. Oncol. 1990, 1, 301–302. [Google Scholar] [CrossRef]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef]
- Prat, A.; Parera, M.; Adamo, B.; Peralta, S.; Perez-Benavente, M.A.; Garcia, A.; Gil-Moreno, A.; Martinez-Palones, J.M.; Baselga, J.; del Campo, J.M. Risk of recurrence during follow-up for optimally treated advanced epithelial ovarian cancer (EOC) with a low-level increase of serum CA-125 levels. Ann. Oncol. 2009, 20, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Chen, Z.; Fadiel, A.; Naftolin, F.; Eichenbaum, K.D.; Xia, Y. Circulation DNA: Biological implications for cancer metastasis and immunology. Med. Hypotheses 2005, 65, 956–961. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Lippman, M.; Osborne, C.K. Circulating tumor DNA--ready for prime time? N. Engl. J. Med. 2013, 368, 1249–1250. [Google Scholar] [CrossRef]
- Figg, W.D.; Reid, J. Monitor tumor burden with circulating tumor DNA. Cancer Biol. Ther. 2013, 14, 697–698. [Google Scholar] [CrossRef]
- Kurniali, P.C.; Storandt, M.H.; Jin, Z. Utilization of Circulating Tumor Cells in the Management of Solid Tumors. J. Pers. Med. 2023, 13, 104483. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Fortina, P. RG Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 369, 93–94. [Google Scholar]
- Wood-Bouwens, C.M.; Haslem, D.; Moulton, B.; Almeda, A.F.; Lee, H.; Heestand, G.M.; Nadauld, L.D.; Ji, H.P. Therapeutic Monitoring of Circulating DNAMutations in Metastatic Cancer with Personalized Digital, P.C.R. J. Mol. Diagn. 2020, 22, 247–261. [Google Scholar] [CrossRef]
- Hayashi, H.; Kunimasa, K.; Tanishima, S.; Nakamura, K.; Ishikawa, M.; Kato, Y.; Aimono, E.; Kawano, R.; Nishihara, H. Germline BRCA2 variant with low variant allele frequency detected in tumor-only comprehensive genomic profiling. Cancer Sci. 2024, 115, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Esteva-Socias, M.; Enver-Sumaya, M.; Gómez-Bellvert, C.; Guillot, M.; Azkárate, A.; Marsé, R.; Sastre, Ú.; Blasco, A.; Calabuig-Fariñas, S.; Asensio, V.J.; et al. Detection of the EGFRG719SMutation in Non-small Cell Lung Cancer Using Droplet Digital, P.C.R. Front. Med. 2020, 7, 594900. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Ding, T.; Tang, D.; Xi, M. The survival outcome and complication of secondary cytoreductive surgery plus chemotherapy in recurrent ovarian cancer: A systematic review and meta-analysis. J. Ovarian Res. 2021, 14, 93. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, L.; Chen, Y.; Qing, C. Circulating cell-free DNA and circulating tumor cells, the “liquid biopsies” in ovarian cancer. J. Ovarian Res. 2017, 10, 75. [Google Scholar] [CrossRef]
- Coetzee, S.G.; Shen, H.C.; Hazelett, D.J.; Lawrenson, K.; Kuchenbaecker, K.; Tyrer, J.; Rhie, S.K.; Levanon, K.; Karst, A.; Drapkin, R.; et al. Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Hum. Mol. Genet. 2015, 24, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Bukłaho, P.A.; Kiśluk, J.; Wasilewska, N.; Nikliński, J. Molecular features as promising biomarkers in ovarian cancer. Adv. Clin. Exp. Med. 2023, 32, 1029–1040. [Google Scholar] [CrossRef]
- Capizzi, E.; Gabusi, E.; Grigioni, A.D.E.; De Iaco, P.; Rosati, M.; Zamagni, C.; Fiorentino, M. Quantification of free plasma DNA before and after chemotherapy in patients with advanced epithelial ovarian cancer. Diagn. Mol. Pathol. 2008, 17, 34–38. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 2014, 6, ra26–ra224. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma, D.N.A. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma, D.N.A. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Otsuka, J.; Okuda, T.; Sekizawa, A.; Amemiya, S.; Saito, H.; Okai, T.; Kushima, M. Detection of p53 mutations in the plasma DNA of patients with ovarian cancer. Int. J. Gynecol. Cancer 2004, 14, 459–464. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification monitoring of cancer mutations by targeted deep sequencing of plasma, D.N.A. Sci. Transl. Med. 2012, 4, ra68–ra136. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, M.; Wohlmuth, C.; Waas, M.; Bernardini, M.Q.; Kislinger, T. High-throughput approaches for precision medicine in high-grade serous ovarian cancer. J. Hematol. Oncol. 2020, 13, 134. [Google Scholar] [CrossRef]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, ra154–ra162. [Google Scholar] [CrossRef] [PubMed]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guo, F.; Bagnoli, M.; Xue, F.X.; Sun, B.C.; Shmulevich, I.; Mezzanzanica, D.; Chen, K.X.; Sood, A.K.; Yang, D.; et al. Key nodes of a microRNA network associated with the integrated mesenchymal subtype of high-grade serous ovarian cancer. Chin. J. Cancer 2015, 34, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Criscitiello, C.; Locatelli, M.; Milano, M.; Curigliano, G. Liquid biopsies for solid tumors: Understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol. Ther. 2016, 157, 120–124. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PATIENT | Age | Sample Time Points | Study Length (Days) | Neo-adyuvance | FIGO | PCI | R Surgery | Relapse | Origin | Exitus | Mutation(s) Identified by NGS | Mutation-Specific ctDNA Detected by dPCR (Líquid Biopsy) | Verified in Tumor by dPCR (Tumor Tissue) | Further NGS-Identified Pathogenic Mutations | Further NGS-Identified Mutations Classified as VUS (Variant of Uncertain Significance) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 61 | 5 | 538 | YES | IIIC | 30 | R1 | YES | Bulgarian | YES | TP53 C135Sfs*35 | ||||
| 2 | 42 | 5 | 391 | YES | IVA | 18 | R1 | YES | Spanish | YES | BRCA2 G2793V | ||||
| 3 | 61 | 6 | 882 | NO | IIIC2 | 2 | R0 | NO | Spanish | NO | |||||
| 4 | 72 | 5 | 401 | YES | IVB | 11 | YES | YES | TP53 P27L | ||||||
| 5 | 70 | 4 | 400 | YES | IIIA2 | 7 | R0 | YES | Spanish | NO | TP53 R175H | + | + | BRCA1 T688* | |
| 6 | 80 | 4 | 384 | YES | IIB | 23 | R0 | YES | Spanish | NO | |||||
| 7 | 70 | 3 | 301 | NO | IVB | 9 | R1 | NO | Spanish | NO | TP53 R175H BRCA1 E1210R | + | + | ||
| 8 | 60 | 6 | 997 | NO | IVB | 18 | R0 | YES | Spanish | NO | TP53 p.? | + | + | ||
| 9 | 63 | 4 | 357 | YES | IIIC | 12 | YES | YES | TP53 A86Hfs*37 BRCA2 K3326* | ||||||
| 10 | 53 | 9 | 933 | YES | IIIA2 | 16 | R0 | NO | Moroccan | NO | TP53 G245D | o | + | BRCA1 G1770V | |
| 11 | 51 | 7 | 812 | YES | IIIC | 21 | R0 | YES | Romanian | NO | BRCA1 E272* TP53 R248Q | + + | + + | ||
| 12 | 75 | 7 | 835 | YES | NA | 35 | R0 | YES | Spanish | NO | |||||
| 13 | 75 | 4 | 356 | NO | IIIC | 13 | R0 | YES | Spanish | NO | TP53 c.911_919+7delCTAAGCGAGGTAAGCA | ||||
| 14 | 51 | 4 | 532 | YES | IIIA | 22 | R0 | NO | Chinese | NO | TP53 Y220C | + | + | BRCA2 S611Yfs*5 | BRCA1 K654R |
| 15 | 53 | 4 | 365 | NO | IC | 10 | R0 | NO | Spanish | NO | BRCA1 Y655* C994* | ||||
| 16 | 74 | 7 | 314 | NO | IIIA | 17 | R0 | NO | Spanish | NO | TP53 R249S | + | + | TP53 R254S | |
| 17 | 64 | 5 | 288 | NO | IIIB | 7 | R0 | NO | Spanish | NO | |||||
| 18 | 59 | 5 | 300 | NO | IIIc | 12 | R0 | NO | Spanish | NO | TP53 C275F | ||||
| 19 | 63 | 5 | 280 | YES | IVa | 19 | R1 | YES | Spanish | NO | BRCA2c.8332-1G>A = p.? | BRCA2 G2270R E2832K S2984L BRCA1 E438K S186F | |||
| 20 | 72 | 4 | 360 | NO | IVa | 15 | R1 | NO | Spanish | NO | BRCA1 S1428* | ||||
| 21 | 70 | 5 | 420 | YES | IIIc | 20 | R0 | NO | Romanian | NO | TP53 Y236* | BRCA2 D1441N BRCA1 P1010S | |||
| 22 | 68 | 5 | 400 | YES | IIIc | 18 | R0 | YES | Moroccan | NO | TP53 H193R | BRCA2 A2851L |
| Cycling Step | Temperature (°C) | Time (s) | Number of Cycles |
|---|---|---|---|
| Enzyme activation | 95 | 600 | 1 |
| Denaturation | 94 | 30 | 40 |
| Annealing/extension | 50–60 * (optimum) | 60 ** | 40 |
| Enzyme deactivation | 98 | 600 | 1 |
| Hold | 4 | Infinite | 1 |
| Patient | Correlation between: | ||||
| cfDNA-TP53 (R175H)ctDNA | cfDNA-CA125 | TP53 (R175H) ctDNA-CA125 | |||
| 5 | 0.53 (p = 0.09) | 0.88 (p < 0.001) | 0.73 (p = 0.011) | ||
| Patient | Correlation between: | ||||
| cfDNA-TP53 R248Q (c.743 G>A) ctDNA | cfDNA- BRCA1 E272* (c.814 G>T) ctDNA | cfDNA-CA125 | TP53 R248Q (c.743 G>A) ctDNA-CA125 | BRCA1 E272* (c.814 G>T) ctDNA-CA125 | |
| 11 | 0.58 (p = 0.001) | 0.59 (p = 0.001) | 0.87 (p < 0.01) | 0.43 (p = 0.02) | 0.52 (p = 0.005) |
| Patient | Correlation between | ||||
| cfDNA-TP53 G245 ctDNA | cfDNA-CA125 | TP53 G245 ctDNA-CA125 | |||
| 10 | 0.93 (p < 0.001) | 0.68 (p < 0.001) | 0.66 (p < 0.001) | ||
| Patient | Correlation between | ||||
| cfDNA-TP53 (c.994C-1G) ctDNA | cfDNA-CA125 | TP53 (c.994C-1G) ctDNA-CA125 | |||
| 8 | 0.59 (p = 0.05) | 0.61 (p = 0.04) | 0.71 (p = 0.01) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llueca, A.; Canete-Mota, S.; Jaureguí, A.; Barneo, M.; Ibañez, M.V.; Neef, A.; Ochoa, E.; Tomas-Perez, S.; Mari-Alexandre, J.; Gilabert-Estelles, J.; et al. The Impact of Liquid Biopsy in Advanced Ovarian Cancer Care. Diagnostics 2024, 14, 1868. https://doi.org/10.3390/diagnostics14171868
Llueca A, Canete-Mota S, Jaureguí A, Barneo M, Ibañez MV, Neef A, Ochoa E, Tomas-Perez S, Mari-Alexandre J, Gilabert-Estelles J, et al. The Impact of Liquid Biopsy in Advanced Ovarian Cancer Care. Diagnostics. 2024; 14(17):1868. https://doi.org/10.3390/diagnostics14171868
Chicago/Turabian StyleLlueca, Antoni, Sarai Canete-Mota, Anna Jaureguí, Manuela Barneo, Maria Victoria Ibañez, Alexander Neef, Enrique Ochoa, Sarai Tomas-Perez, Josep Mari-Alexandre, Juan Gilabert-Estelles, and et al. 2024. "The Impact of Liquid Biopsy in Advanced Ovarian Cancer Care" Diagnostics 14, no. 17: 1868. https://doi.org/10.3390/diagnostics14171868
APA StyleLlueca, A., Canete-Mota, S., Jaureguí, A., Barneo, M., Ibañez, M. V., Neef, A., Ochoa, E., Tomas-Perez, S., Mari-Alexandre, J., Gilabert-Estelles, J., Serra, A., Climent, M. T., Bellido, C., Ruiz, N., Segarra-Vidal, B., & Llueca, M. (2024). The Impact of Liquid Biopsy in Advanced Ovarian Cancer Care. Diagnostics, 14(17), 1868. https://doi.org/10.3390/diagnostics14171868