
Identifying the Pathogenic Variants in Heart Genes in Vietnamese Sudden Unexplained Death Victims by Next-Generation Sequencing

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Targeted Next-Generation Sequencing
2.3. Sanger Sequencing
2.4. In Silico Prediction
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winkel, B.G.; Holst, A.G.; Theilade, J.; Kristensen, I.B.; Thomsen, J.L.; Ottesen, G.L.; Bundgaard, H.; Svendsen, J.H.; Haunsø, S.; Tfelt-Hansen, J. Nationwide Study of Sudden Cardiac Death in Persons Aged 1–35 Years. Eur. Heart J. 2011, 32, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the Europea. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Wong, C.X.; Brown, A.; Lau, D.H.; Chugh, S.S.; Albert, C.M.; Kalman, J.M.; Sanders, P. Epidemiology of Sudden Cardiac Death: Global and Regional Perspectives. Heart Lung Circ. 2019, 28, 6–14. [Google Scholar] [CrossRef]
- Wisten, A.; Forsberg, H.; Krantz, P.; Messner, T. Sudden cardiac death in 15–35-year olds in Sweden during 1992–99. J. Intern. Med. 2002, 252, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Puranik, R.; Chow, C.K.; Duflou, J.A.; Kilborn, M.J.; McGuire, M.A. Sudden Death in the Young. Heart Rhythm 2005, 2, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Eckart, R.E.; Shry, E.A.; Burke, A.P.; McNear, J.A.; Appel, D.A.; Castillo-Rojas, L.M.; Avedissian, L.; Pearse, L.A.; Potter, R.N.; Tremaine, L.; et al. Sudden Death in Young Adults: An Autopsy-Based Series of a Population Undergoing Active Surveillance. J. Am. Coll. Cardiol. 2011, 58, 1254–1261. [Google Scholar] [CrossRef]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac Channel Molecular Autopsy: Insights from 173 Consecutive Cases of Autopsy-negative Sudden Unexplained Death Referred for Postmortem Genetic Testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef]
- Christiansen, S.L.; Hertz, C.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; Camp, L.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Genetic Investigation of 100 Heart Genes in Sudden Unexplained Death Victims in a Forensic Setting. Eur. J. Hum. Genet. 2016, 24, 1797–1802. [Google Scholar] [CrossRef]
- Huynh, M.T.; Proust, A.; Bouligand, J.; Popescu, E. AKAP9-Related Channelopathy: Novel Pathogenic Variant and Review of the Literature. Genes 2022, 13, 2167. [Google Scholar] [CrossRef]
- Wu, J.; Ding, W.G.; Horie, M. Molecular Pathogenesis of Long QT Syndrome Type 1. J. Arrhythmia 2016, 32, 381–388. [Google Scholar] [CrossRef]
- Hedley, P.L.; Jorgensen, P.; Schlamowitz, S.; Wangari, R.; Moolman-Smook, J.; Brink, P.A.; Kanters, J.K.; Corfield, V.A.; Christiansen, M. The Genetic Basis of Long QT and Short QT Syndromes: A Mutation Update. Hum. Mutat. 2009, 30, 1486–1511. [Google Scholar] [CrossRef]
- Tester, D.J.; Ackerman, M.J. Cardiomyopathic and Channelopathic Causes of Sudden Unexplained Death in Infants and Children. Annu. Rev. Med. 2009, 60, 69–84. [Google Scholar] [CrossRef]
- Wijeyeratne, Y.D.; Tanck, M.W.; Mizusawa, Y.; Batchvarov, V.; Barc, J.; Crotti, L.; Bos, J.M.; Tester, D.J.; Muir, A.; Veltmann, C.; et al. SCN5A Mutation Type and a Genetic Risk Score Associate Variably With Brugada Syndrome Phenotype in SCN5A Families. Circ. Genom. Precis. Med. 2020, 13, E002911. [Google Scholar] [CrossRef] [PubMed]
- Sacilotto, L.; Scanavacca, M.I.; Olivetti, N.; Lemes, C.; Pessente, G.D.; Wulkan, F.; Hachul, D.T.; Krieger, J.E.; Pereira, A.C.; Darrieux, F.C.C. Low Rate of Life-Threatening Events and Limitations in Predicting Invasive and Noninvasive Markers of Symptoms in a Cohort of Type 1 Brugada Syndrome Patients: Data and Insights From the GenBra Registry. J. Cardiovasc. Electrophysiol. 2020, 31, 2920–2928. [Google Scholar] [CrossRef] [PubMed]
- Di Resta, C.; Berg, J.; Villatore, A.; Maia, M.; Pili, G.; Fioravanti, F.; Tomaiuolo, R.; Sala, S.; Benedetti, S.; Peretto, G. Concealed Substrates in Brugada Syndrome: Isolated Channelopathy or Associated Cardiomyopathy? Genes 2022, 13, 1755. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick Eduardo, B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. Expert Consensus Statement on the state of genetic testing for cardiac diseases. J. Arrhythmia 2022, 38, 491–553. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Denjoy, I.; Extramiana, F.; Maltret, A.; Buisson, N.R.; Lupoglazoff, J.M.; Klug, D.; Hayashi, M.; Takatsuki, S.; Villain, E.; et al. Incidence and Risk Factors of Arrhythmic Events in Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2009, 119, 2426–2434. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Bhuiyan, Z.A.; Tester, D.J.; Hofman, N.; Bikker, H.; van Tintelen, J.P.; Mannens, M.M.; Wilde, A.A.; Ackerman, M.J. The RYR2-encoded Ryanodine Receptor/calcium Release Channel in Patients Diagnosed Previously with Either Catecholaminergic Polymorphic Ventricular Tachycardia or Genotype Negative, Exercise-induced Long QT Syndrome: A Comprehensive Open Reading Frame Mutational Analysis. J. Am. Coll. Cardiol. 2009, 54, 2065–2074. [Google Scholar]
- Priori, S.G.; Chen, S.R. Inherited Dysfunction of Sarcoplasmic Reticulum Ca2+ Handling and Arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A Missense Mutation in a Highly Conserved Region of CASQ2 is Associated with Autosomal Recessive Catecholamine-induced Polymorphic Ventricular Tachycardia in Bedouin Families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef]
- Nyegaard, M.; Overgaard, M.T.; Søndergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in Calmodulin Cause Ventricular Tachycardia and Sudden Cardiac Death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of Triadin, a Protein of the Calcium Release Complex, is Responsible for Cardiac Arrhythmia with Sudden Death in Human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef]
- Asatryan, B.; Medeiros-Domingo, A. Molecular and Genetic Insights into Progressive Cardiac Conduction Disease. Europace 2019, 21, 1145–1158. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Tan, B.; Crotti, L.; Tester, D.J.; Eckhardt, L.; Cuoretti, A.; Kroboth, S.L.; Song, C.; Zhou, Q.; Kopp, D.; et al. Gain-of-function Mutation S422L in the KCNJ8-encoded Cardiac K(ATP) Channel Kir6.1 as a Pathogenic Substrate for J-wave Syndromes. Heart Rhythm 2010, 7, 1466–1471. [Google Scholar] [CrossRef]
- Noseworthy, P.; Tikkanen, J.; Porthan, K.; Oikarinen, L.; Pietila, A.; Harald, K.; Peloso, G.M.; Merchant, F.M.; Jula, A.; Väänänen, H.; et al. The Early Repolarization Pattern in the General Population: Clinical Correlates and Heritability. J. Am. Coll. Cardiol. 2011, 57, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, W.; Kaess, B.; Debiec, R.; Nelson, C.P.; Stark, K.; Tobin, M.D.; Macfarlane, P.W.; Tomaszewski, M.; Samani, N.J.; Hengstenberg, C. Heritability of Early Repolarization: A Population-based Study. Circ. Cardiovasc. Genet. 2011, 4, 134–138. [Google Scholar] [CrossRef]
- Watanabe, H.; Nogami, A.; Ohkubo, K.; Kawata, H.; Hayashi, Y.; Ishikawa, T.; Makiyama, T.; Nagao, S.; Yagihara, N.; Takehara, N.; et al. Electrocardiographic Characteristics and SCN5A Mutations in Idiopathic Ventricular Fibrillation Associated with Early Repolarization. Circ. Arrhythmia Electrophysiol. 2011, 4, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 is a Novel Brugada and Early Repolarization Syndrome Susceptibility Gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef]
- Takayama, K.; Ohno, S.; Ding, W.; Ashihara, T.; Fukumoto, D.; Wada, Y.; Makiyama, T.; Kise, H.; Hoshiai, M.; Matsuura, H.; et al. A de novo Gain-of-function KCND3 Mutation in Early Repolarization Syndrome. Heart Rhythm 2019, 16, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L. Cerebral Small Vessel Disease: From Pathogenesis and Clinical Characteristics to Therapeutic Challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Mancuso, M.; Arnold, M.; Bersano, A.; Burlina, A.; Chabriat, H.; Debette, S.; Enzinger, C.; Federico, A.; Filla, A.; Finsterer, J.; et al. Monogenic Cerebral Smallvessel Diseases: Diagnosis and Therapy. Consensus Recommendations of the European Academy of Neurology. Eur. J. Neurol. 2020, 27, 909–927. [Google Scholar]
- Ullal, A.J.; Abdelfattah, R.S.; Ashley, E.A.; Froelicher, V.F. Hypertrophic Cardiomyopathy as a Cause of Sudden Cardiac Death in the Young: A Meta-analysis. Am. J. Med. 2016, 129, 486–496. [Google Scholar] [CrossRef]
- Veselka, J.; Anavekar, N.S.; Charron, P. Hypertrophic Obstructive Cardiomyopathy. Lancet 2017, 389, 1253–1267. [Google Scholar] [CrossRef]
- Abbas, M.T.; Baba Ali, N.; Farina, J.M.; Mahmoud, A.K.; Pereyra, M.; Scalia, I.G.; Kamel, M.A.; Barry, T.; Lester, S.J.; Cannan, C.R.; et al. Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future. Biomedicines 2024, 12, 682. [Google Scholar] [CrossRef]
- Bonaventura, J.; Polakova, E.; Vejtasova, V.; Veselka, J. Genetic Testing in Patients with Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 10401. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef]
- Miura, A.; Yamamoto, T.; Funayama, K.; Koyama, A.; Takatsuka, H.; Sato, T.; Nishio, H. Postmortem Identification of Genetic Variations Associated with Sudden Unexpected Death in Young People. Int. Heart J. 2024, 65, 55–62. [Google Scholar] [CrossRef]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Fordyce, S.L.; Dahl, M.; Holst, A.G.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation Sequencing of 34 Genes in Sudden Unexplained Death Victims in Forensics and in Patients with Channelopathic Cardiac Diseases. Int. J. Legal Med. 2015, 129, 793–800. [Google Scholar] [CrossRef]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; LuCamp, P.E.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation Sequencing of 100 Candidate Genes in Young Victims of Suspected Sudden Cardiac Death with Structural Abnormalities of the Heart. Int. J. Legal Med. 2016, 130, 91–102. [Google Scholar] [CrossRef]
- Castiglione, V.; Modena, M.; Aimo, A.; Chiti, E.; Botto, N.; Vittorini, S.; Guidi, B.; Vergaro, G.; Barison, A.; Rossi, A.; et al. Molecular Autopsy of Sudden Cardiac Death in the Genomics Era. Diagnostics 2021, 11, 1378. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hedley, P.L.; Jørgensen, P.; Schlamowitz, S.; Moolman-Smook, J.; Kanters, J.K.; Corfield, V.A.; Christiansen, M. The Genetic Basis of Brugada Syndrome: A Mutation Update. Hum. Mutat. 2009, 30, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Jipin Das, K.; Duflou, J.; Semsarian, C. Exome Analysis–Based Molecular Autopsy in Cases of Sudden Unexplained Death in the Young. Heart Rhythm 2014, 11, 655–662. [Google Scholar] [CrossRef]
- Hata, Y.; Kinoshita, K.; Mizumaki, K.; Yamaguchi, Y.; Hirono, K.; Ichida, F.; Takasaki, A.; Mori, H.; Nishida, N. Postmortem Genetic Analysis of Sudden Unexplained Death Syndrome under 50 Years of Age: A next-Generation Sequencing Study. Heart Rhythm 2016, 13, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Modena, M.; Castiglione, V.; Aretini, P.; Mazzanti, C.M.; Chiti, E.; Giannoni, A.; Emdin, M.; Di Paolo, M. Unveiling a Sudden Unexplained Death Case by Whole Exome Sequencing and Bioinformatic Analysis. Mol. Genet. Genom. Med. 2020, 8, e1182. [Google Scholar] [CrossRef]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated Disc Abnormalities, Reduced Na(+) Current Density, and Conduction Slowing in Desmoglein-2 Mutant Mice Prior to Cardiomyopathic Changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef]
- Zhang, M.; Tavora, F.; Oliveira, J.B.; Li, L.; Franco, M.; Fowler, D.; Zhao, Z.; Burke, A. PKP2 Mutations in Sudden Death From Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) and Sudden Unexpected Death with Negative Autopsy (SUDNA). Circ. J. 2012, 76, 189–194. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, C.; Rao, F.; Modi, R.M.; Zhu, J.; Liu, X.; Mai, L.; Tan, H.; Yu, X.; Lin, Q.; et al. Silencing of Desmoplakin Decreases Connexin 43/Nav1.5 Expression and Sodium Current in HL1 Cardiomyocytes. Mol. Med. Rep. 2013, 8, 780–786. [Google Scholar]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Gusky, H.C.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate with a Brugada Syndrome Phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Lahrouchi, N.; Priori, S.G. Genetics of Sudden Cardiac Death. Circ. Res. 2015, 116, 1919–1936. [Google Scholar] [CrossRef]
- Cerrone, M.; Priori, S.G. Genetics of Sudden Death: Focus on Inherited Channelopathies. Eur. Heart J. 2011, 32, 2109–2118. [Google Scholar] [CrossRef]
- Tester, D.J.; Spoon, D.B.; Valdivia, H.H.; Makielski, J.C.; Ackerman, M.J. Targeted Mutational Analysis of the RyR2-encoded Cardiac Ryanodine Receptor in Sudden Unexplained Death: A Molecular Autopsy of 49 Medical Examiner/coroner’s Cases. Mayo Clin. Proc. 2004, 79, 1380–1384. [Google Scholar] [CrossRef]
- Chen, L.; Marquardt, L.M.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase Anchoring Protein Causes Long-QT Syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef]
- Maltese, E.P.; Orlova, N.; Krasikova, E.; Emelyanchik, E.; Cheremisina, A.; Kuscaeva, A.; Salmina, A.; Miotto, R.; Bonizzato, A.; Guerri, G.; et al. Gene-targeted Analysis of Clinically Diagnosed Long QT Russian Families. Int. Heart J. 2017, 58, 81–87. [Google Scholar] [CrossRef]
- Bottigliero, D.; Monaco, I.; Santacroce, R.; Casavecchia, G.; Correale, M.; Guastafierro, F.; Leccese, A.; Cordisco, G.; Leva, R.; Trunzo, R.; et al. Novel AKAP9 Mutation and Long QT Syndrome in a Patient with Torsade de Pointes. J. Interv. Card. Electrophysiol. 2019, 56, 171–172. [Google Scholar] [CrossRef]
- Tse, G.; Lee, S.; Zhou, J.; Liu, T.; Wong, C.K.I.; Mak, C.; Mok, N.S.; Jeevaratnam, K.; Zhang, Q.; Cheng, S.H.; et al. Territory-wide Chinese Cohort of Long QT Syndrome: Random Survival Forest and Cox Analyses. Front. Cardiovasc. Med. 2021, 8, 608592. [Google Scholar] [CrossRef]
- Allegue, C.; Coll, M.; Mates, J.; Campuzano, O.; Iglesias, A.; Sobrino, B.; Brion, M.; Amigo, J.; Carracedo, A.; Brugada, P.; et al. Genetic Analysis of Arrythmogenic Disease in the Era of NGS: The Complexity of Clinical Decision-Making in Brugada Syndrome. PLoS ONE 2015, 10, e0133037. [Google Scholar] [CrossRef]
- Garris, R.; Vasudev, R.; Gupta, P.; Tiyyagura, S.; Shamoon, F.; Bikkina, M. Brugada Syndrome and AKAP9: Reconciling Clinical Findings with Diagnostic Uncertainty. J. Electrocardiol. 2019, 57, 119–121. [Google Scholar] [CrossRef]
- Campuzano, O.; Allegue, C.; Sarquella-Brugada, G.; Coll, M.; Mates, J.; Alcalde, M.; Ferrer-Costa, C.; Iglesias, A.; Brugada, J.; Brugada, R. The Role of Clinical, Genetic and Segregation Evaluation in Sudden Infant Death. Forensic Sci. Int. 2014, 242, 9–15. [Google Scholar] [CrossRef]
- Neubauer, J.; Lecca, R.M.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Exome Analysis in 34 Sudden Unexplained Death (SUD) Victims Mainly Identified Variants in Channelopathy-associated Genes. Int. J. Legal Med. 2018, 132, 1057–1065. [Google Scholar] [CrossRef]
- Li, J.L.; Wang, B.Y.; Qu, F.P.; Ma, L.; Liu, K.; Yang, L.; Nie, J.S.; Xi, M.Y.; Jia, L.P.; Tang, X.; et al. Genetic Analysis of Yunnan Sudden Unexplained Death by Whole Exome Sequencing in Southwest of China. J. Forensic Leg. Med. 2020, 70, 101896. [Google Scholar] [CrossRef]
- Jaouadi, H.; Bouyacoub, Y.; Chabrak, S.; Kraoua, L.; Zaroui, A.; Elouej, S.; Nagara, M.; Dallali, H.; Delague, V.; Levy, N.; et al. Multiallelic Rare Variants Support an Oligogenic Origin of Sudden Cardiac Death in the Young. Herz 2021, 46, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.V.; Duff, H.; Gerull, B.; Sumner, G. Early Repolarization Syndrome: A Case Report Focusing on Dynamic Electrocardiographic Changes Before Ventricular Arrhythmias and Genetic Analysis. Heart Rhythm Case Rep. 2015, 1, 213–216. [Google Scholar] [CrossRef]
- Qui, H.; Li, W.H.; Zhang, H.S.; Zhou, G.X.; Li, P.W. Torsades de Pointes Episode in a Woman with High-grade Fever and Inflammatory Activation: A Case Report. World J. Clin. Cases 2021, 9, 2899–2907. [Google Scholar]
- Forleo, C.; D’Erchia, M.A.; Sorrentino, S.; Manzari, C.; Chiara, M.; Iacoviello, M.; Guaricci, I.A.; Santis, D.D.; Musci, L.R.; La Spada, A.; et al. Targeted Next-Generation Sequencing Detects Novel Gene-phenotype Associations and Expands the Mutational Spectrum in Cardiomyopathies. PLoS ONE 2017, 12, e0181842. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, K.; Dam, V.S.; Kjaer-Sorensen, K.; Pedersen, L.N.; Arvydas Skeberdis, V.; Jurevičius, J.; Treinys, R.; Petersen, I.M.B.S.; Nielsen, M.S.; Oxvig, C.; et al. Loss-of-activity-mutation in the Cardiac Chloride-bicarbonate Exchanger AE3 Causes Short QT Syndrome. Nat. Commun. 2017, 8, 1696. [Google Scholar] [CrossRef]
- Christiansen, M.K.; Kjær-Sørensen, K.; Clavsen, N.C.; Dittmann, S.; Jensen, M.F.; Guldbrandsen, H.O.; Pedersen, L.N.; Sørensen, R.H.; Lildballe, D.L.; Müller, K.; et al. Genetic Analysis Identifies the SLC4A3 Anion Exchanger as a Major Gene for Short QT Syndrome. Heart Rhythm 2023, 20, 1136–1143. [Google Scholar] [CrossRef]
- Bianchi, S.; Di Palma, C.; Gallus, G.N.; Taglia, I.; Poggiani, A.; Rosini, F.; Rufa, A.; Muresanu, D.F.; Cerase, A.; Dotti, M.T.; et al. Two Novel HTRA1 Mutations in a European CARASIL Patient. Neurology 2014, 82, 898–900. [Google Scholar] [CrossRef]
- Hara, K.; Shiga, A.; Fukutake, T.; Nozaki, H.; Miyashita, A.; Yokoseki, A.; Kawata, H.; Koyama, A.; Arima, K.; Takahashi, T.; et al. Association of HTRA1 Mutations and Familial Ischemic Cerebral Small-vessel Disease. N. Engl. J. Med. 2009, 360, 1729–1739. [Google Scholar] [CrossRef]
- Ito, J.; Nozaki, H.; Toyoshima, Y.; Abe, T.; Sato, A.; Hashidate, H.; Igarashi, S.; Onodera, O.; Takahashi, H.; Kakita, A. Histopathologic Features of an Autopsied Patient with Cerebral Small Vessel Disease and a Heterozygous HTRA1 Mutation. Neuropathology 2018, 38, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Bougea, A.; Velonakis, G.; Spantideas, N.; Anagnostou, E.; Paraskevas, G.; Kapaki, E.; Kararizou, E. The First Greek Case of Heterozygous Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: An Atypical Clinico-radiological Presentation. Neuroradiol. J. 2017, 30, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Favaretto, S.; Margoni, M.; Salviati, L.; Pianese, L.; Manara, R.; Baracchini, C. A New Italian Family with HTRA1 Mutation Associated with Autosomal-dominant Variant of CARASIL: Are We Pointing Towards a Disease Spectrum? J. Neurol. Sci. 2019, 396, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Nie, S.; Li, Z.; Zhang, Z.; Xiao, T. A New Chinese Family of Autosomal Dominantly inherited Cerebral Small Vessel Disease Related to A Heterozygous HTRA1 Mutation. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Nozaki, H.; Kato, T.; Nihonmatsu, M.; Saito, Y.; Mizuta, I.; Noda, T.; Koike, R.; Miyazaki, K.; Kaito, M.; Ito, S.; et al. Distinct Molecular Mechanisms of HTRA1 Mutants in Manifesting Heterozygotes with CARASIL. Neurology 2016, 86, 1964–1974. [Google Scholar] [CrossRef] [PubMed]

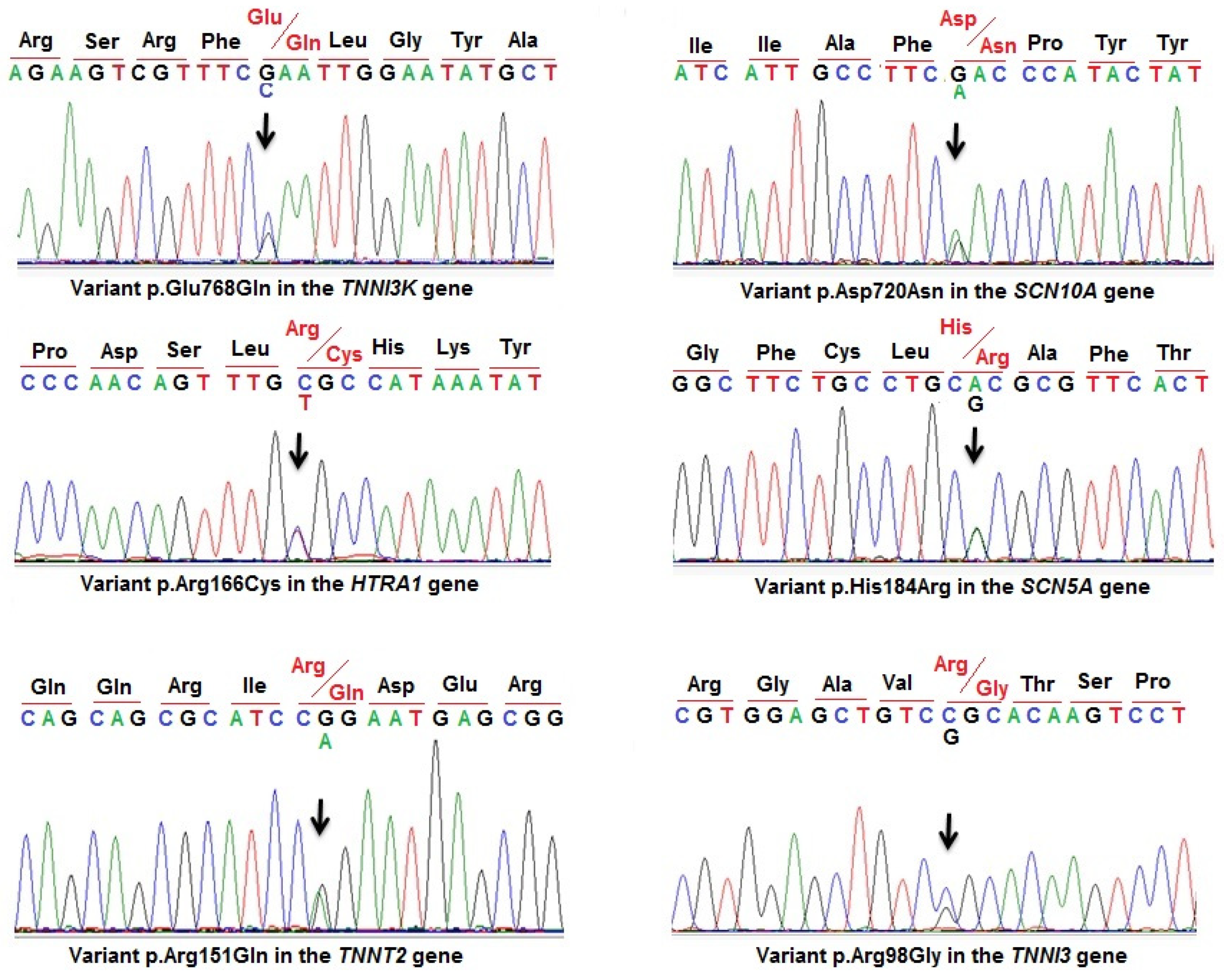

{kind=link}
{kind=link}
| ID | Sex/Age | Event at SUD | Medical History | Autopsy Findings | |
|---|---|---|---|---|---|
| Height (cm) | Health | ||||
| P1 | F/14 | Sleep at night | 160 | Normal | Negative |
| P2 | M/29 | Sleep at night | 178 | Normal | Negative |
| P3 | M/25 | Driving/daytime | 158 | Normal | Negative |
| P4 | M/19 | Sleep at night | 175 | Normal | Negative |
| P5 | M/28 | Sleep at night | 174 | Normal | Negative |
| P6 | M/40 | Eating at night | 152 | Normal | Negative |
| P7 | M/39 | Sleep at daytime | 170 | Normal | Negative |
| P8 | M/22 | Rest at daytime | 162 | Normal | Negative |
| P9 | M/29 | Sleep at night | 168 | Normal | Negative |
| P10 | F/33 | Sleep at night | 158 | Normal | Negative |
| P11 | F/25 | Rest at daytime | 156 | Normal | Negative |
| P12 | M/35 | Sleep at night | 170 | Normal | Negative |
| P13 | M/39 | Sleep at night | 165 | Normal | Negative |
| P14 | M/24 | Working/daytime | 180 | Normal | Negative |
| P15 | M/23 | Sleep at night | 168 | Normal | Negative |
| P16 | M/27 | Working/daytime | 160 | Normal | Negative |
| P17 | M/30 | Working/daytime | 172 | Normal | Negative |
| P18 | F/1 | Rest at daytime | 76 | Normal | Negative |
| P19 | M/30 | Sleep at night | 169 | Normal | Negative |
| P20 | M/40 | Sleep at night | 172 | Normal | Negative |
| P21 | F/32 | Rest at daytime | 159 | Normal | Negative |
| P22 | M/27 | Working/daytime | 160 | Normal | Negative |
| P23 | M/34 | Sleep at night | 167 | Normal | Negative |
| P24 | M/40 | Sleep at night | 162 | Normal | Negative |
| P25 | M/38 | Sleep at night | 170 | Normal | Negative |
| P26 | M/40 | Rest at daytime | 162 | Normal | Negative |
| P27 | M/35 | Sleep at daytime | 164 | Normal | Negative |
| P28 | F/2 | Rest at daytime | 80 | Normal | Negative |
| P29 | M/33 | Rest at daytime | 162 | Normal | Negative |
| P30 | M/27 | Sleep at night | 173 | Normal | Negative |
| P31 | M/35 | Sleep at night | 170 | Normal | Negative |
| P32 | M/32 | Sleep at night | 162 | Normal | Negative |
| P33 | M/33 | Sleep at night | 167 | Normal | Negative |
| P34 | M/39 | Playing a sport | 166 | Normal | Negative |
| P35 | M/24 | Sleep at night | 179 | Normal | Negative |
| P36 | M/19 | Working/daytime | 169 | Normal | Negative |
| P37 | F/29 | Rest at daytime | 155 | Normal | Negative |
| P38 | F/20 | Sleep at night | 158 | Normal | Negative |
| P39 | M/23 | Sleep at night | 168 | Normal | Negative |
| P40 | F/40 | Sleep at night | 159 | Normal | Negative |
| ID | Gene | cDNA/Protein | dbSNP/MAF/ClinVar/ExAC | Zygosity |
|---|---|---|---|---|
| P1 (F/14) | RYR2 (NM_001035.3) | c.51C>G p.Phe17Leu | novel | het |
| P2 (M/29) | AKAP9 (NM_005751.4) | c.5187_5188dup p.Arg1730llefsTer4 | novel | het |
| P3 (M/25) | TNNI3K (NM_015978.3) | c.2302G>C p.Glu768Gln | rs202238194/0.00000 RCV000768402.5/pathogenic | het |
| P4 (M/19) | KCNA5 (NM_002234.4) | c.683C>A p.Pro228His | VCV002202820.2 uncertain significance | het |
| P5 (M/28) | MYBPC3 (NM_000256.3) | c.2275G>A p.Glu759Lys | rs750810342/0.00002487 VCV000843772.16/uncertain significance | het |
| P6 (M/40) | HTRA1 (NM_002775.5) | c.496C>T p.Arg166Cys | rs2097494390/VCV001325819.5 RCV002291765.4/pathogenic | het |
| P7 (M/39) | MYH6 (NM_002471.4) | c.1454A>T p.Lys485Met | novel | het |
| P8 (M/22) | TNNT2 (NM_001276345.2) | c.452G>A p.Arg151Gln | rs730881101/0.00000 RCV000796707.6/pathogenic | het |
| P9 (M/29) | SCN10A (NM_006514.4) | c.2158G>A p.Asp720Asn | rs781354273/0.00006/VCV000532067.8 pathogenic | het |
| P10 (F/33) | CSRP3 (NM_003476.5) | c.298C>T p.Arg100Cys | rs201214593/0.00004/VCV000851709.9 uncertain significance | het |
| P11 (F/25) | MYLK (NM_053025.4) | c.4840G>A p.Glu1614Lys | novel | het |
| AKAP9 (NM_005751.4) | c.9215G>T p.Gly3072Val | novel | het | |
| P12 (M/35) | SCN5A (NM_00335.5) | c.515A>G p.His184Arg | rs794728898/0.000102/VCV000201540.5 likely pathogenic | het |
| P13 (M/39) | TNNI3 (NM_000363.5) | c.292C>G p.Arg98Gly | rs730881068/0.00005/VCV001331910.2 uncertain significance | het |
| P14 (M/24) | GSN (NM_198252.3) | c.872T>C p.Ile291Thr | novel | het |
| P15 (M/23) | SLC4A3 (NM_005070.4) | c.2535+1G>A | novel | het |
| LAMA4 (NM_001105206.3) | c.5025A>T p.Glu1675Asp | novel | het |
| (A) | |||||||
|---|---|---|---|---|---|---|---|
| Gene/Variants | ClinVar | CADD Score/Prediction | FATHMM Score/Prediction | Mutation Taster Score/Prediction | PhD-SNP Score/Prediction | Polyphen 2 Score/Prediction | SNP&GO Score/Prediction |
| RYR2 c.10498G>T p.Asp3500Tyr | Novel | 25.5 Probably damaging | −4.21 Damaging | 160 Deleterious | Neutral RI8 | 1.000 Possibly damaging | Disease RI1 |
| KCNA5 c.683C>A p.Pro228His | Uncertain significance | 24.1 Probably damaging | −0.21 Tolerated | 77 Deleterious | Neutral RI5 | 1.000 Possibly damaging | Disease RI7 |
| MYBPC3 c.2275G>A p.Glu759Lys | Uncertain significance | 26.7 Probably damaging | −0.20 Tolerated | 56 Deleterious | Neutral RI5 | 1.000 Possibly damaging | - |
| MYH6 c.1454A>T p.Lys485Met | Novel | 28.9 Probably damaging | −2.60 Damaging | - | Disease RI6 | 1.000 Possibly damaging | Disease RI9 |
| CSRP3 c.298C>T p.Arg100Cys | Uncertain significance | 28.5 Probably damaging | −0.28 Tolerated | 89 Benign | - | 0.110 Benign | Disease RI2 |
| MYLK c.4840G>A p.Glu1614Lys | Novel | 36.0 Probably damaging | - | - | Neutral RI2 | 0.611 Possibly damaging | - |
| AKAP9 c.9215G>T p.Gly3072Val | Novel | 17.6 Tolerated | 3.76 Tolerated | - | Neutral RI8 | 0.973 Possibly damaging | - |
| TNNI3 c.292C>G p.Arg98Gly | Uncertain significance | 26.3 Probably damaging | −3.49 Damaging | 125 Deleterious | Neutral RI1 | 1.000 Possibly damaging | Disease RI10 |
| GSN c.872T>C p.Ile291Thr | Novel | 29.1 Probably damaging | −0.34 Tolerated | Benign | Neutral RI7 | 1.000 Possibly damaging | - |
| LAMA4 c.5025A>T p.Glu1675Asp | Novel | 23.6 Probably damaging | −1.32 Tolerated | 21 Benign | Disease RI4 | 0.998 Possibly damaging | Neutral RI7 |
| (B) | |||||||
| In Silico Prediction Tools | Wildtype | Mutant | Prediction | ||||
| EX-SKIP | −218.867 | Exon-skipping | |||||
| Fruitfly | NA | NA | - | ||||
| MaxEntScan | 1.51 | −6.67 | Damage variant | ||||
| NetGene2 | 0.60 | 0.00 | Donor loss | ||||
| Spliceailookup | 0.98 | 0.00 | Donor loss | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen Tat, T.; Lien, N.T.K.; Luu Sy, H.; Ta Van, T.; Dang Viet, D.; Nguyen Thi, H.; Tung, N.V.; Thanh, L.T.; Xuan, N.T.; Hoang, N.H. Identifying the Pathogenic Variants in Heart Genes in Vietnamese Sudden Unexplained Death Victims by Next-Generation Sequencing. Diagnostics 2024, 14, 1876. https://doi.org/10.3390/diagnostics14171876
Nguyen Tat T, Lien NTK, Luu Sy H, Ta Van T, Dang Viet D, Nguyen Thi H, Tung NV, Thanh LT, Xuan NT, Hoang NH. Identifying the Pathogenic Variants in Heart Genes in Vietnamese Sudden Unexplained Death Victims by Next-Generation Sequencing. Diagnostics. 2024; 14(17):1876. https://doi.org/10.3390/diagnostics14171876
Chicago/Turabian StyleNguyen Tat, Tho, Nguyen Thi Kim Lien, Hung Luu Sy, To Ta Van, Duc Dang Viet, Hoa Nguyen Thi, Nguyen Van Tung, Le Tat Thanh, Nguyen Thi Xuan, and Nguyen Huy Hoang. 2024. "Identifying the Pathogenic Variants in Heart Genes in Vietnamese Sudden Unexplained Death Victims by Next-Generation Sequencing" Diagnostics 14, no. 17: 1876. https://doi.org/10.3390/diagnostics14171876