
Whole-Genome Omics Elucidates the Role of CCM1 and Progesterone in Cerebral Cavernous Malformations within CmPn Networks

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Treatment, Sample Preparations, and Data Acquisition
2.1.1. Cell Culture and Treatment
2.1.2. Cell Collection, Protein Sample Preparations, and Data Generation
2.1.3. Proteomic Data Acquisition and Processing
2.1.4. RNAseq Data Acquisition
2.2. Omics, Bioinformatics Analysis, and Systems Biology
2.2.1. Pathway Enrichment Analysis
2.2.2. Systems Biology Analysis
2.2.3. ML-Aided Transcriptional Factors (TF) Prediction Analysis
3. Results
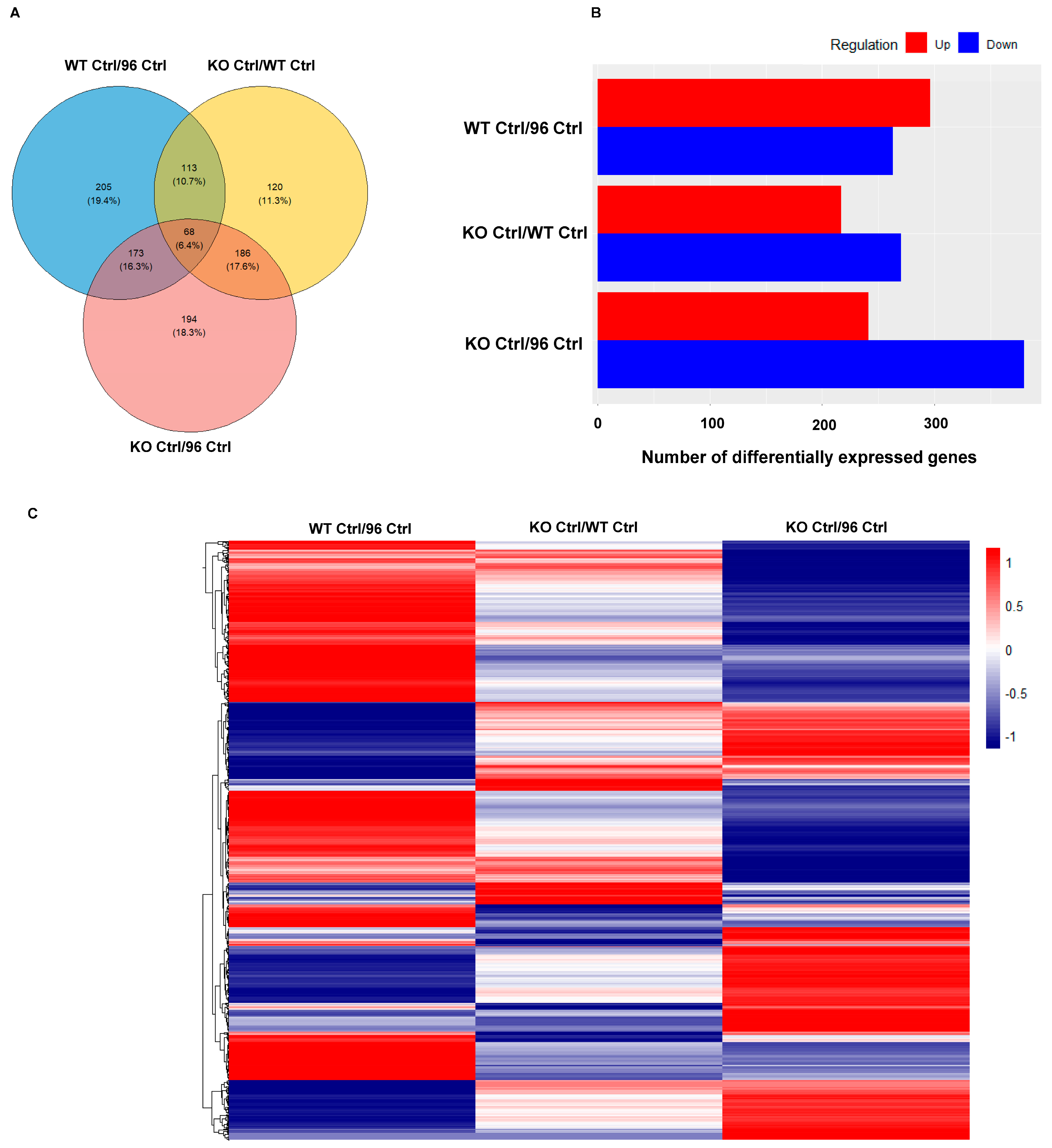
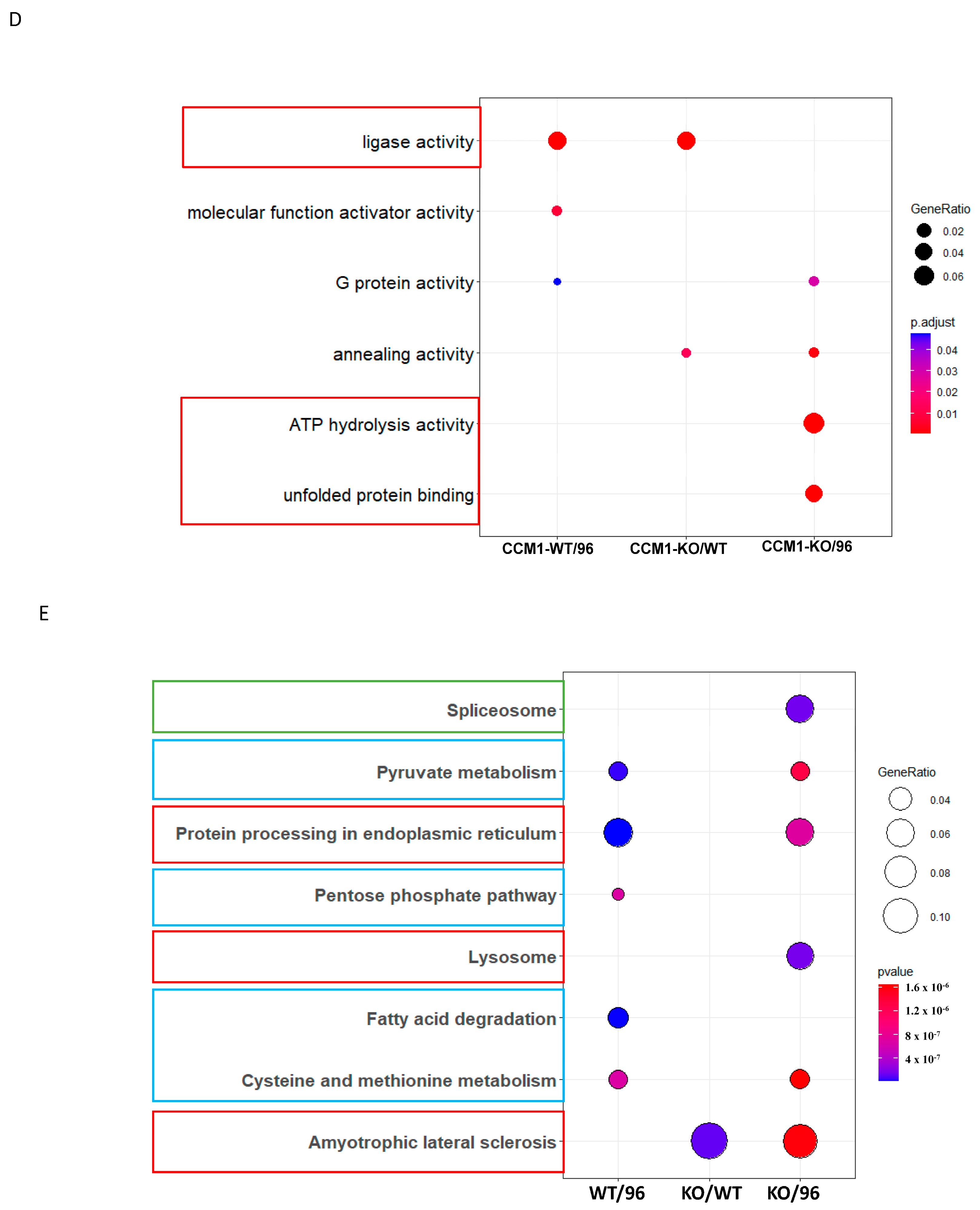
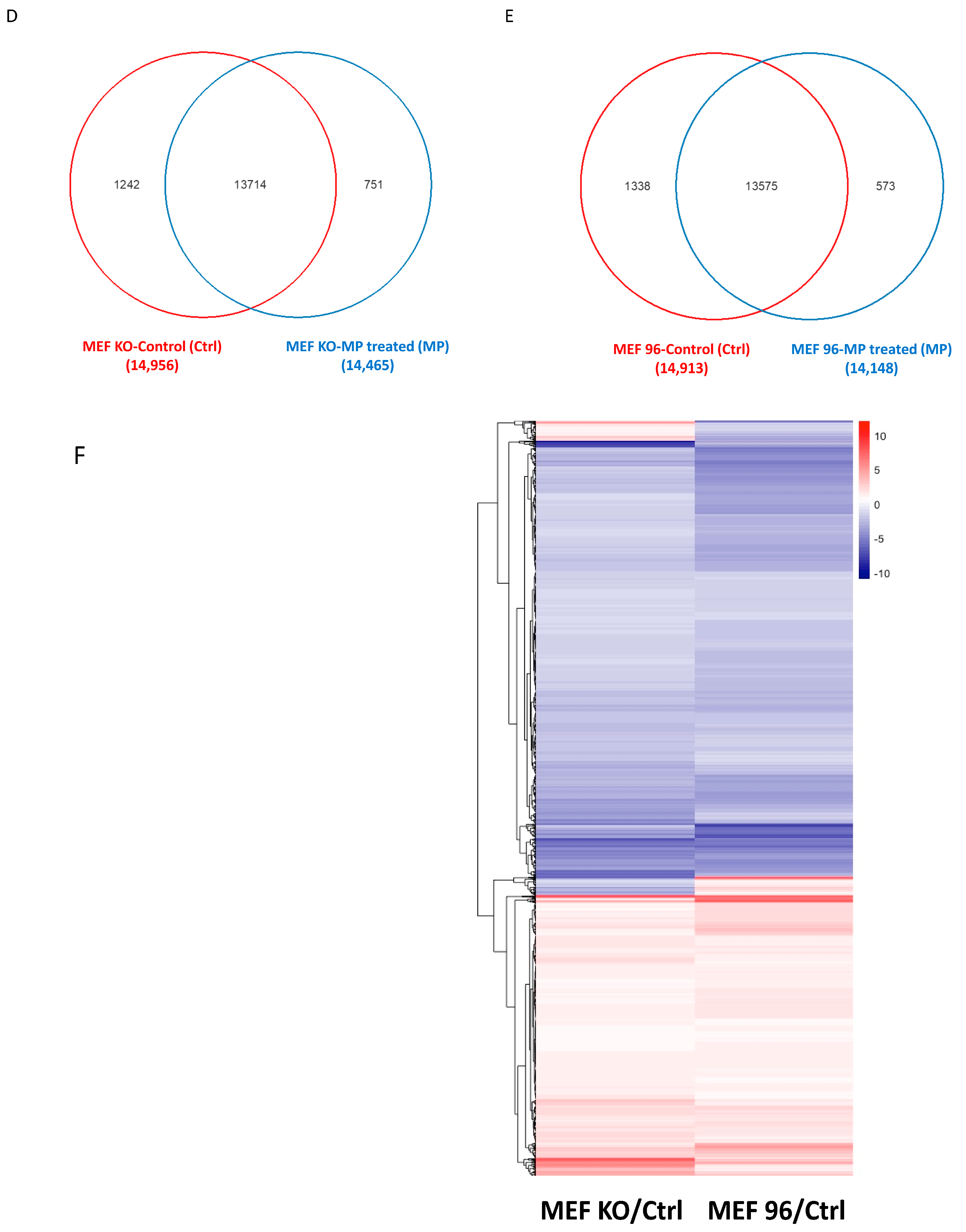
3.1. Differential Expressed Proteins between Mouse Embryonic Fibroblasts (MEFs) with Different CCM1 Genotypes
3.1.1. Differentially Expressed Protein (DEP) Profiles in the MEFs with Different CCM1 Genotypes
3.1.2. Differential Signal Pathways in the MEFs with Different CCM1 Expression Levels
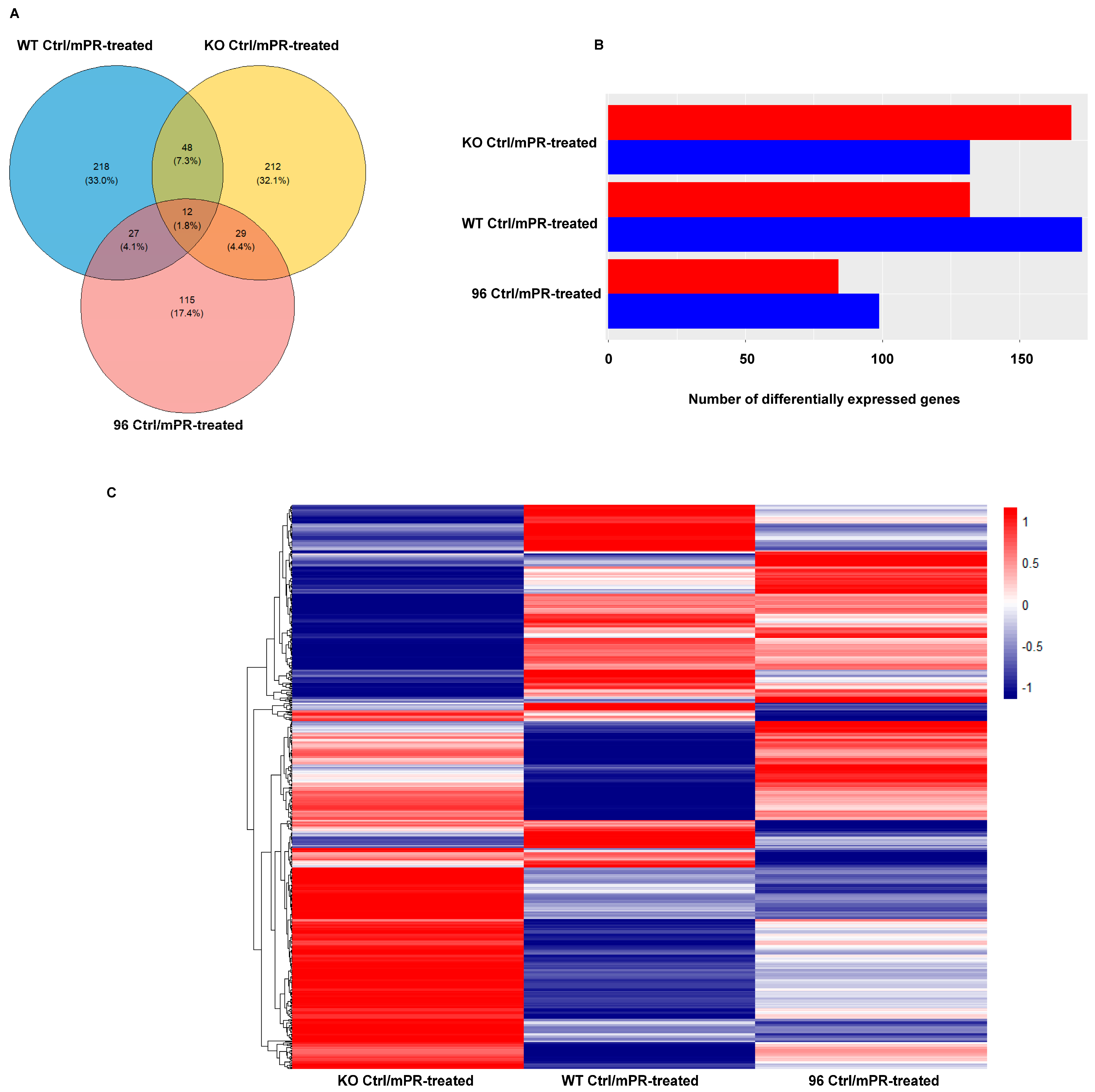
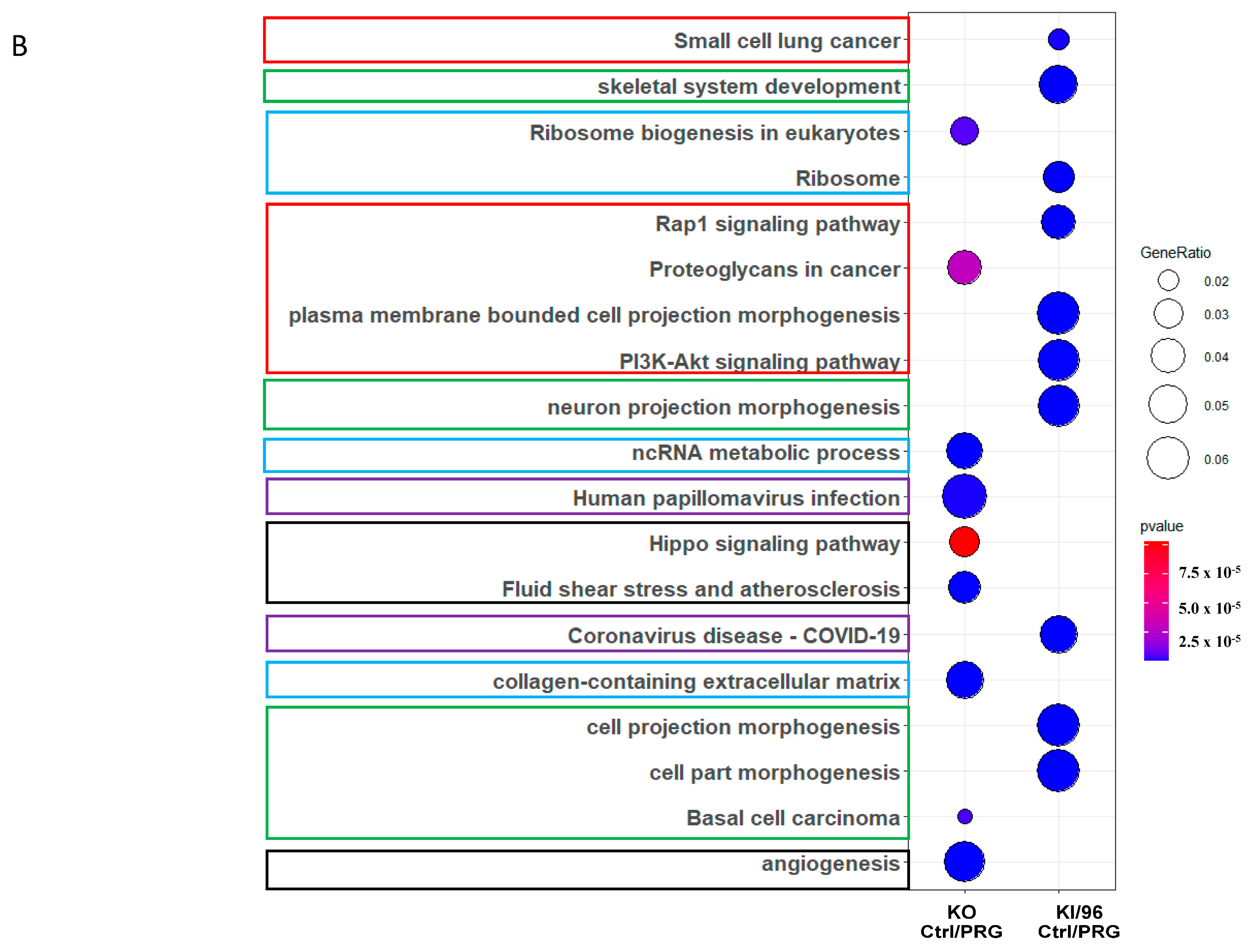
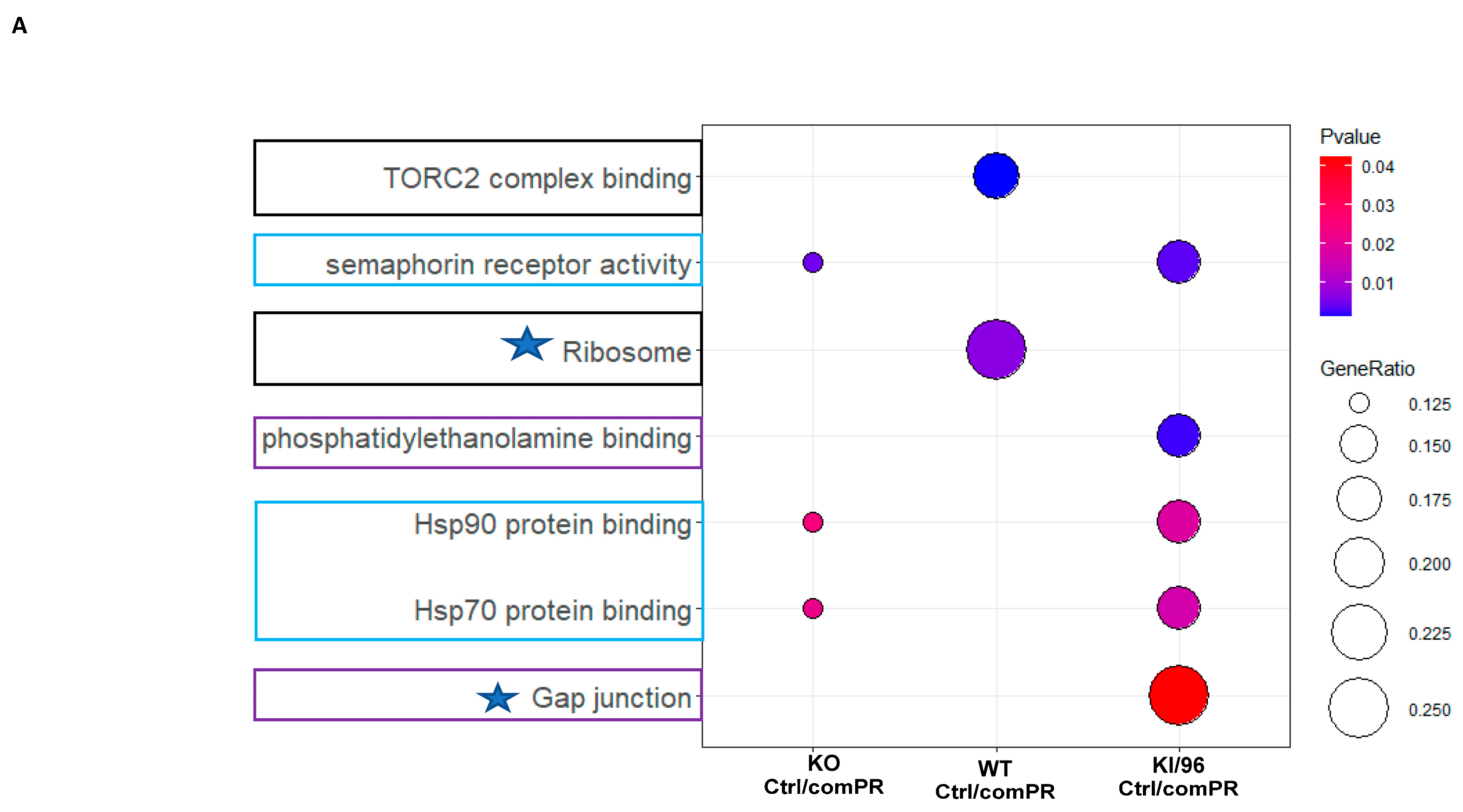
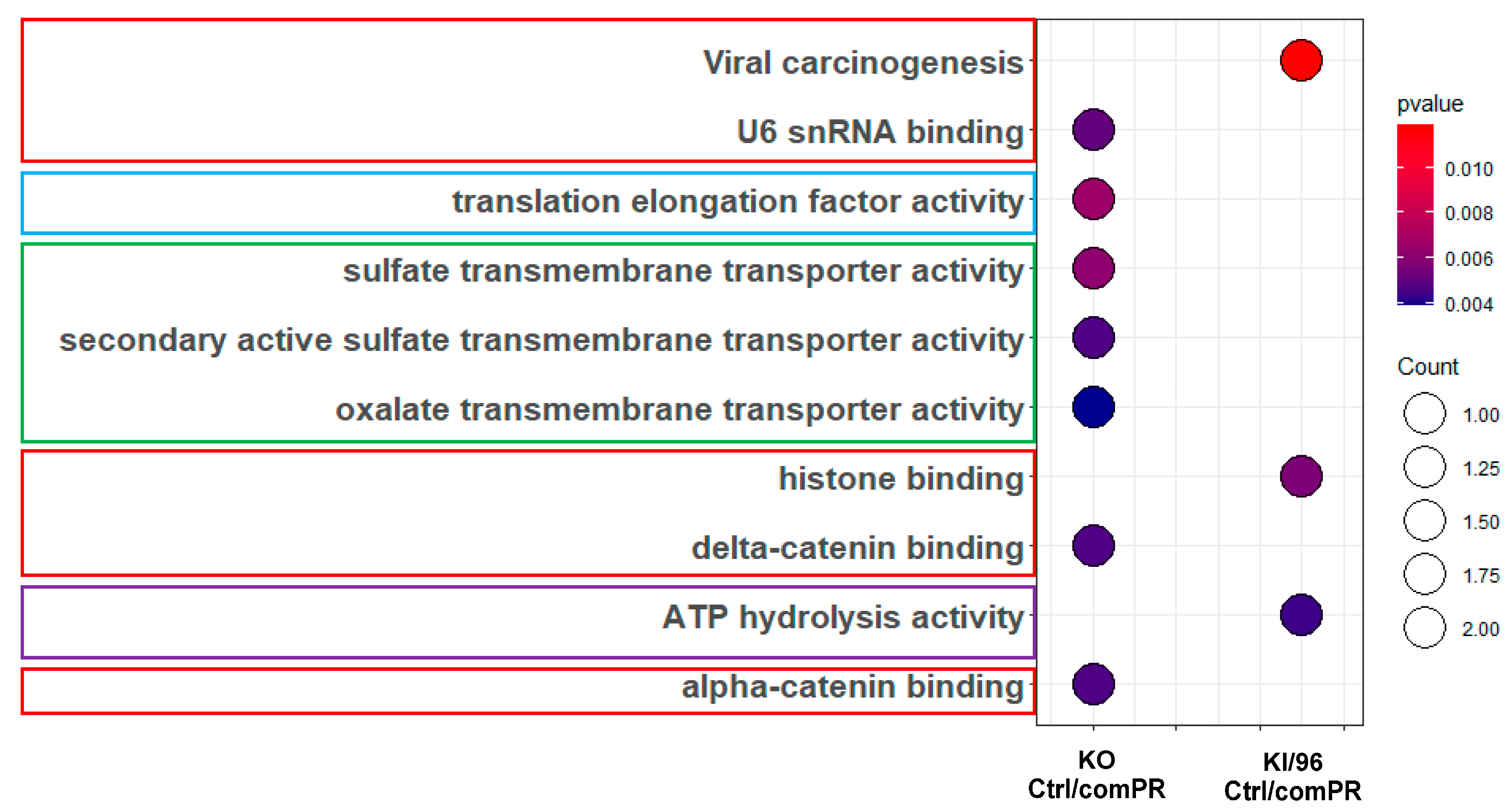
3.2. Differential Expressed Proteins between Mouse Embryonic Fibroblasts with Different CCM1 Genotypes under Progesterone (PRG) Actions
3.2.1. Differentially Expressed Protein (DEP) Profiles in the MEFs with Different CCM1 Genotypes in Response to PRG-Specific Actions
3.2.2. Different Pathway Response in the MEFs with Three Different CCM1 Genotypes under Progesterone (PRG) Actions
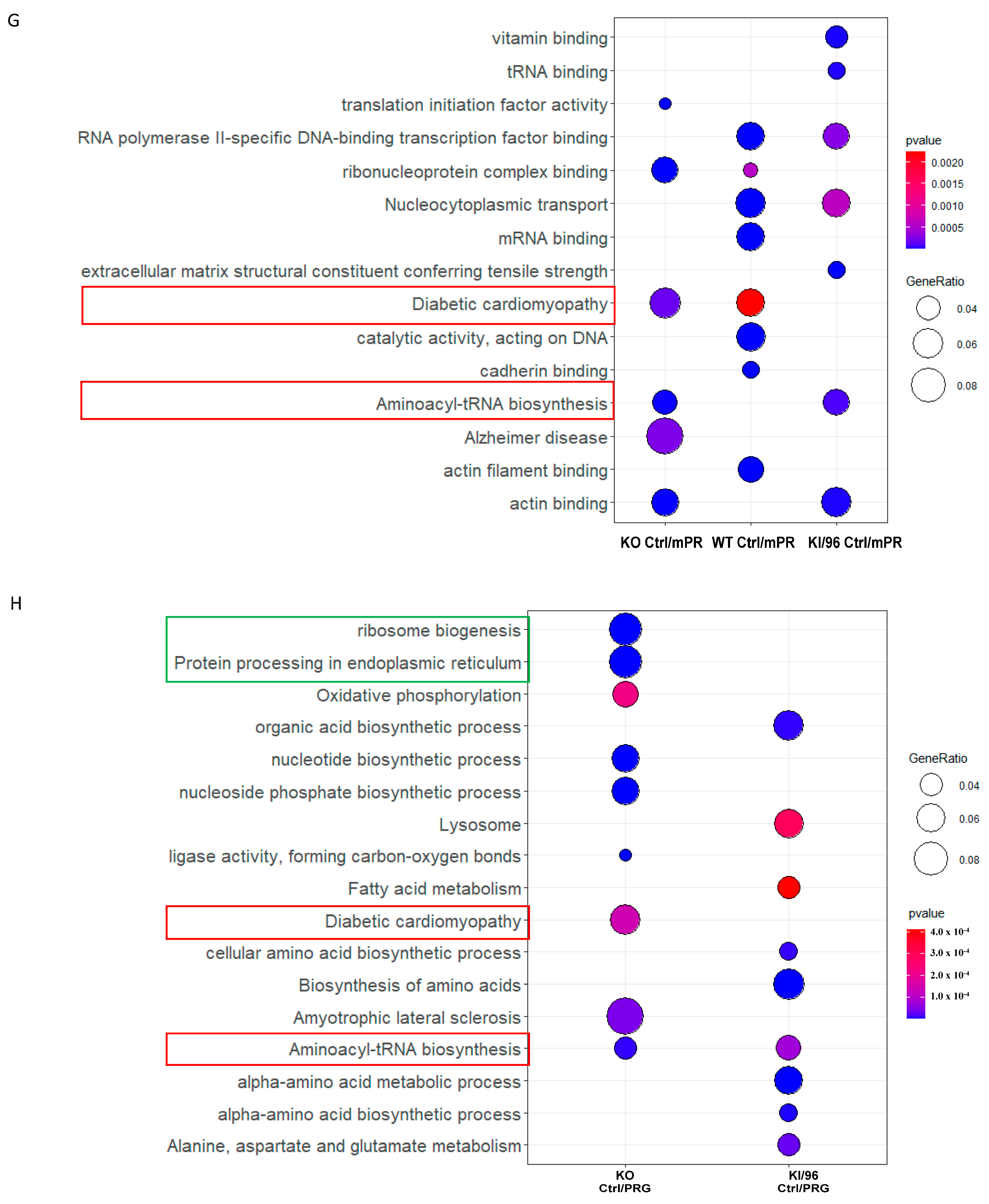
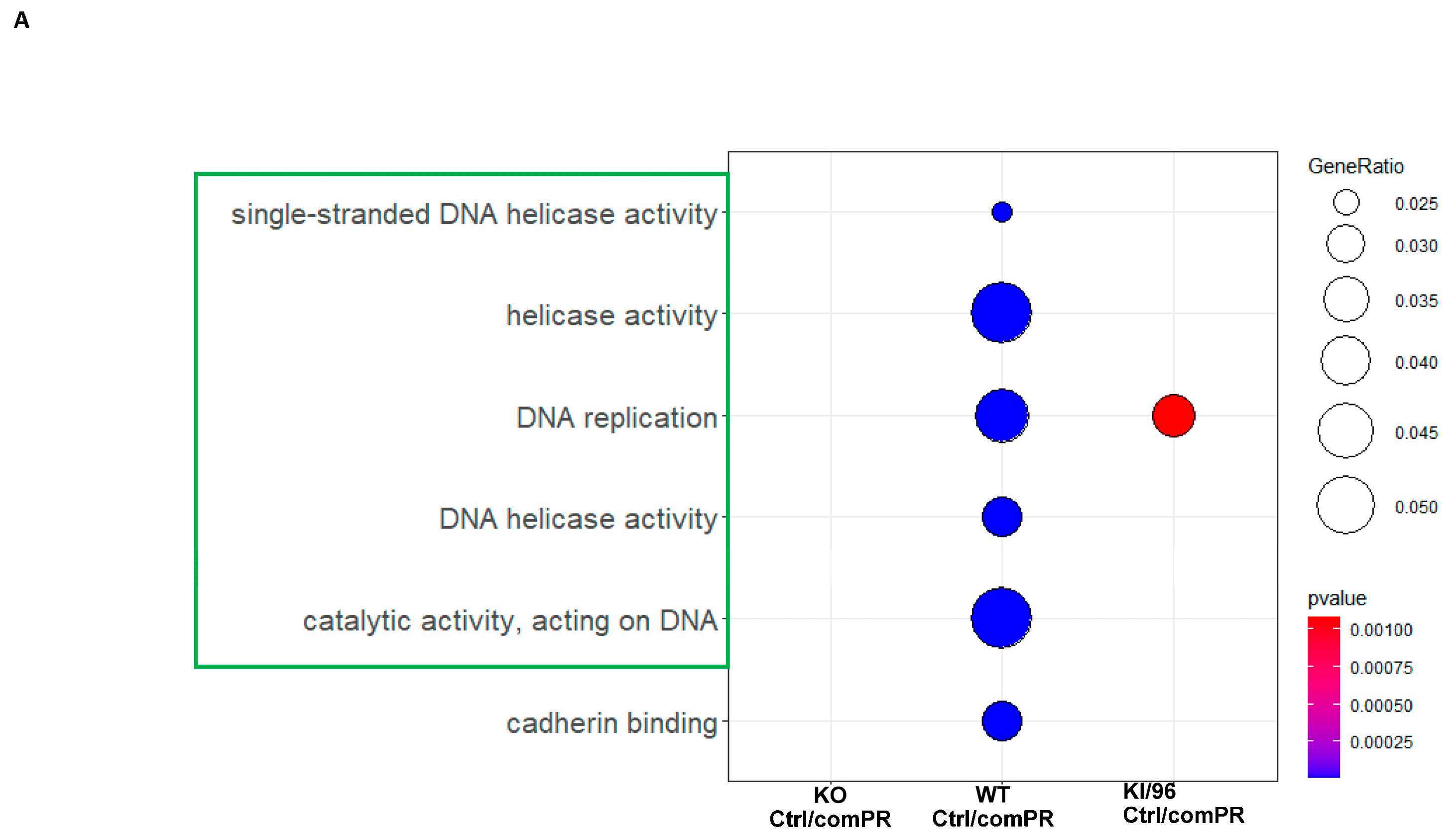
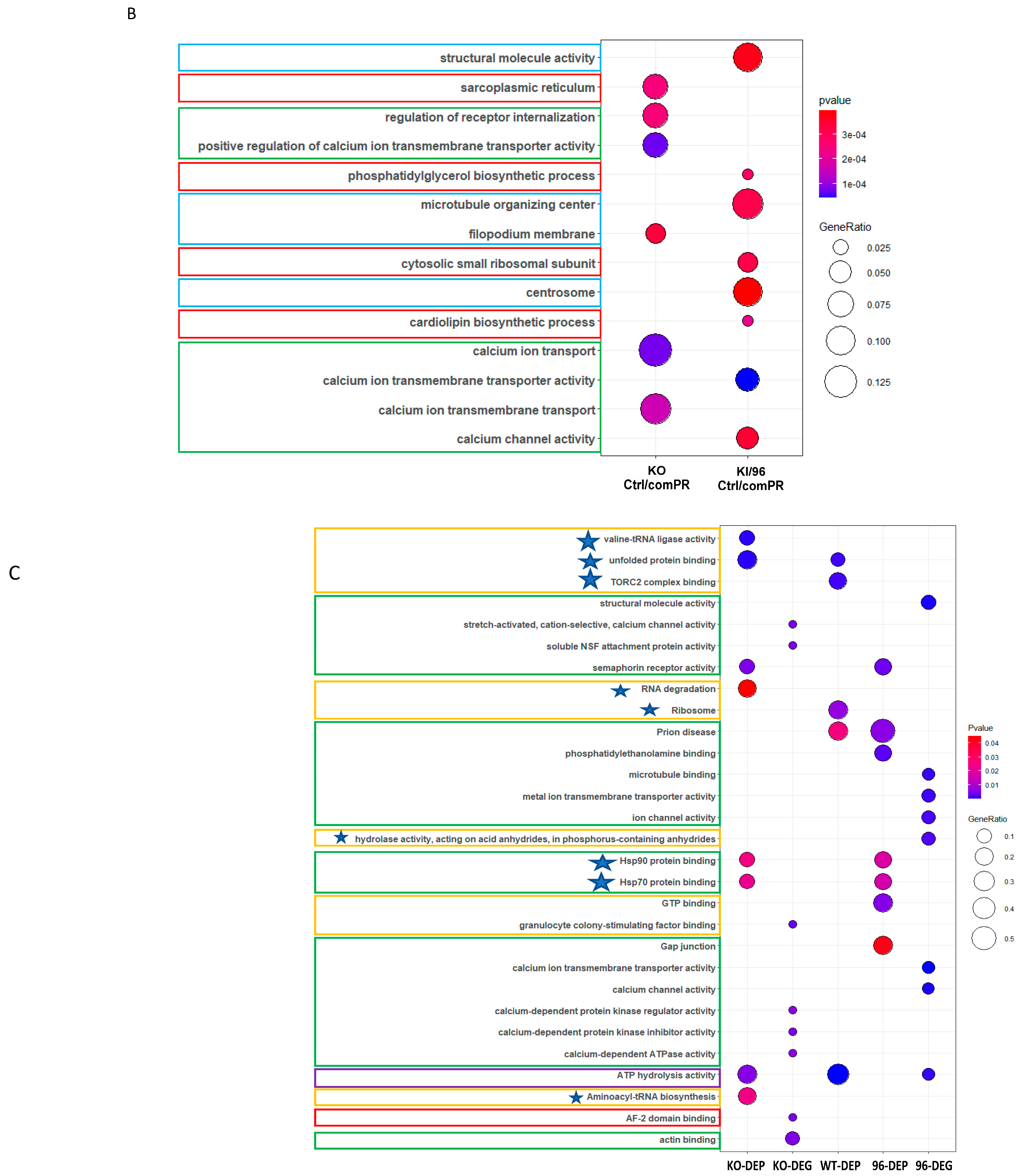
3.3. Pathways Related to Non-mPR PRG Actions Modulated through CCM1 Proteins within CmPn Network Can Be Eliminated by Using Established Proteomic Data
3.4. Identification of mPR-Specific PRG Pathways Modulated through CCM1 Proteins in the CmPn Signal Network
3.4.1. Proteomic Identification of mPR-Specific PRG Pathways Regulated by CCM1 Proteins
3.4.2. Transcriptional Profiling of mPR-Specific PRG Pathways Regulated by CCM1 Proteins
3.4.3. Omics Analysis of mPR-Specific PRG Pathways by CCM1 Proteins
3.5. Discovery of Novel Transcription Factors in mPR-Specific PRG Pathways Regulated by CCM1 in the CmPn Signaling Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Padarti, A.; Zhang, J. Recent advances in cerebral cavernous malformation research. Vessel Plus 2018, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Tournier-Lasserve, E. Molecular genetic screening of CCM patients: An overview. Methods Mol. Biol. 2020, 2152, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, V.; Canzoneri, R.; Perrelli, A.; Arduino, C.; Zonta, A.; Brusco, A.; Retta, S.F. Next-generation sequencing advances the genetic diagnosis of cerebral cavernous malformation (CCM). Antioxidants 2022, 11, 1294. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; Balzac, F.; Degani, S.; Degan, P.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; De Luca, E.; Braggion, S.; Trapani, E.; Guglielmotto, M.; Biasi, F.; Forni, M.; Moglia, A.; Trabalzini, L.; Retta, S.F. KRIT1 loss of function causes a ros-dependent upregulation of c-jun. Free Radic. Biol. Med. 2014, 68, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Trapani, E.; Monache, S.D.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for cerebral cavernous malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Trapani, E.; Monache, S.D.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief. 2018, 16, 929–938. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, P.V.; Kuebel, J.M.; Sarelius, I.H.; Glading, A.J. KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 2014, 289, 33054–33065. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Czupalla, C.J.; Wolburg, H. Current concepts of blood-brain barrier development. Int. J. Dev. Biol. 2011, 55, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood-brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef] [PubMed]
- Salvador, E.; Shityakov, S.; Forster, C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. 2014, 355, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.L.; Kosman, D.J. Aberrant cerebral iron trafficking co-morbid with chronic inflammation: Molecular mechanisms and pharmacologic intervention. Front. Neurol. 2022, 13, 855751. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Witt, K.A.; Hom, S.; Egleton, R.D.; Mark, K.S.; Davis, T.P. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1241–H1248. [Google Scholar] [CrossRef] [PubMed]
- Praestholm, S.M.; Correia, C.M.; Grontved, L. Multifaceted control of gr signaling and its impact on hepatic transcriptional networks and metabolism. Front. Endocrinol. 2020, 11, 572981. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, D.F.; Thankaraj, D.; Jones-Diette, J.; Morgan, R.; Ifesemen, O.S.; Shenker, N.G.; Walsh, D.A. The efficacy of systemic glucocorticosteroids for pain in rheumatoid arthritis: A systematic literature review and meta-analysis. Rheumatology 2021, 61, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.C.; Ott, J. The dialectic role of progesterone. Maturitas 2009, 62, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, S.A.; Boonyaratanakornkit, V.; Edwards, D.P. Progesterone receptor transcription and non-transcription signaling mechanisms. Steroids 2003, 68, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Simoncini, T. Non-genomic sex steroid actions in the vascular system. Semin. Reprod. Med. 2007, 25, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; Hamilton, N.; Marquez-Garban, D.C.; Pateetin, P.; McGowan, E.M.; Pietras, R.J. Extranuclear signaling by sex steroid receptors and clinical implications in breast cancer. Mol. Cell. Endocrinol. 2018, 466, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; Edwards, D.P. Receptor mechanisms mediating non-genomic actions of sex steroids. Semin. Reprod. Med. 2007, 25, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; Edwards, D.P. Receptor mechanisms of rapid extranuclear signalling initiated by steroid hormones. Essays Biochem. 2004, 40, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; Bi, Y.; Rudd, M.; Edwards, D.P. The role and mechanism of progesterone receptor activation of extra-nuclear signaling pathways in regulating gene transcription and cell cycle progression. Steroids 2008, 73, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Renteria, M.; Belkin, O.; Jang, D.; Aickareth, J.; Bhalli, M.; Zhang, J. Cmpn signaling networks in the tumorigenesis of breast cancer. Front. Endocrinol. 2022, 13, 1013892. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Vasquez, M.; Grajeda, B.; Ellis, C.; Zhang, J. Systems-wide analysis unravels the new roles of CCM signal complex (CSC). Heliyon 2019, 5, e02899. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Jiang, X.; Grajeda, B.; Padarti, A.; Ellis, C.C.; Flores, E.; La, O.A.C.-D.; Zhang, J. Ccm signaling complex (CSC) couples both classic and non-classic progesterone receptor signaling. Cell Commun. Signal. 2022, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Bhalli, M.; Grajeda, B.; Zhang, J. Cmp signaling network leads to identification of prognostic biomarkers for triple-negative breast cancer in caucasian women. Genet. Test. Mol. Biomark. 2022, 26, 198–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Abou-Fadel, J. Calm the raging hormone—A new therapeutic strategy involving progesterone-signaling for hemorrhagic CCMs. Vessel Plus 2021, 5, 23. [Google Scholar] [CrossRef]
- Goitre, L.; Fornelli, C.; Zotta, A.; Perrelli, A.; Retta, S.F. Production of KRIT1-knockout and KRIT1-knockin mouse embryonic fibroblasts as cellular models of CCM disease. Methods Mol. Biol. 2020, 2152, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Smith, M.; Falahati, K.; Zhang, J. Comparative omics of CCM signaling complex (CSC). Chin. Neurosurg. J. 2020, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. Upset: Visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Timmerman, E.; Staes, A.; Vuylsteke, M.; De Muynck, L.; Gevaert, K. Protein interactors of 3-o sulfated heparan sulfates in human mci and age-matched control cerebrospinal fluid. Sci. Data 2023, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 2017, 18, 287. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.M.; Liu, J.S.; Guo, D.M.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Cao, P.; Zhao, D.; Zaïane, O.R. An optimized cost-sensitive svm for imbalanced data learning. In Pacific-Asia Conference on Knowledge Discovery and Data Mining; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; Fontanella, M.; Hart, B.L.; Morrison, L.A.; Pawlikowska, L.; Kim, H.; et al. Cytochrome p450 and matrix metalloproteinase genetic modifiers of disease severity in cerebral cavernous malformation type 1. Free Radic. Biol. Med. 2016, 92, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Konate, M.M.; Li, M.C.; McShane, L.M.; Zhao, Y. Discovery of pathway-independent protein signatures associated with clinical outcome in human cancer cohorts. Sci. Rep. 2022, 12, 19283. [Google Scholar] [CrossRef] [PubMed]
- Mubeen, S.; Hoyt, C.T.; Gemund, A.; Hofmann-Apitius, M.; Frohlich, H.; Domingo-Fernandez, D. The impact of pathway database choice on statistical enrichment analysis and predictive modeling. Front. Genet. 2019, 10, 1203. [Google Scholar] [CrossRef] [PubMed]
- von der Ahe, D.; Janich, S.; Scheidereit, C.; Renkawitz, R.; Schutz, G.; Beato, M. Glucocorticoid and progesterone receptors bind to the same sites in two hormonally regulated promoters. Nature 1985, 313, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Neef, G.; Beier, S.; Elger, W.; Henderson, D.; Wiechert, R. New steroids with antiprogestational and antiglucocorticoid activities. Steroids 1984, 44, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; O’Malley, B.W. Expanding functional diversity of the coactivators. Trends Biochem. Sci. 2005, 30, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Oomen, C.A.; Mayer, J.L.; de Kloet, E.R.; Joels, M.; Lucassen, P.J. Brief treatment with the glucocorticoid receptor antagonist mifepristone normalizes the reduction in neurogenesis after chronic stress. Eur. J. Neurosci. 2007, 26, 3395–3401. [Google Scholar] [CrossRef] [PubMed]
- Morgan, F.H.; Laufgraben, M.J. Mifepristone for management of cushing’s syndrome. Pharmacotherapy 2013, 33, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Castinetti, F.; Conte-Devolx, B.; Brue, T. Medical treatment of cushing’s syndrome: Glucocorticoid receptor antagonists and mifepristone. Neuroendocrinology 2010, 92 (Suppl. S1), 125–130. [Google Scholar] [CrossRef] [PubMed]
- Castinetti, F.; Brue, T.; Conte-Devolx, B. The use of the glucocorticoid receptor antagonist mifepristone in cushing’s syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.T.; Chang, C.W.; Wei, R.J.; Wang, S.N. Progesterone and related compounds in hepatocellular carcinoma: Basic and clinical aspects. Biomed Res. Int. 2013, 2013, 290575. [Google Scholar] [CrossRef] [PubMed]
- P’Eng, F.K.; Lui, W.Y.; Chang, T.J.; Kao, H.L.; Wu, L.H.; Liu, T.Y.; Chi, C.W. Glucocorticoid receptors in hepatocellular carcinoma and adjacent liver tissue. Cancer 1988, 62, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Leehy, K.A.; Anderson, T.M.R.; Daniel, A.R.; Lange, C.A.; Ostrander, J.H. Modifications to glucocorticoid and progesterone receptors alter cell fate in breast cancer. J. Mol. Endocrinol. 2016, 56, R99–R114. [Google Scholar] [CrossRef] [PubMed]
- Ogara, M.F.; Rodriguez-Segui, S.A.; Marini, M.; Nacht, A.S.; Stortz, M.; Levi, V.; Presman, D.M.; Vicent, G.P.; Pecci, A. The glucocorticoid receptor interferes with progesterone receptor-dependent genomic regulation in breast cancer cells. Nucleic Acids Res. 2019, 47, 10645–10661. [Google Scholar] [CrossRef] [PubMed]
- Honer, C.; Nam, K.; Fink, C.; Marshall, P.; Ksander, G.; Chatelain, R.E.; Cornell, W.; Steele, R.; Schweitzer, R.; Schumacher, C. Glucocorticoid receptor antagonism by cyproterone acetate and ru486. Mol. Pharmacol. 2003, 63, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Reid, V.; Le, A.; Croft, J.; Zhang, J. Key members of the cmpn as biomarkers distinguish histological and immune subtypes of hepatic cancers. Diagnostics 2023, 13, 1012. [Google Scholar] [CrossRef] [PubMed]
- Aickareth, J.; Hawwar, M.; Sanchez, N.; Gnanasekaran, R.; Zhang, J. Membrane progesterone receptors (mprs/paqrs) are going beyond its initial definitions. Membranes 2023, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Grajeda, B.; Jiang, X.; La, O.A.C.-D.; Flores, E.; Padarti, A.; Bhalli, M.; Le, A.; Zhang, J. Cmp signaling network unveils novel biomarkers for triple negative breast cancer in African American women. Cancer Biomark. 2022, 34, 607–636. [Google Scholar] [CrossRef] [PubMed]
- Renteria, M.; Belkin, O.; Aickareth, J.; Jang, D.; Hawwar, M.; Zhang, J. Zinc’s association with the cmpn/cmp signaling network in breast cancer tumorigenesis. Biomolecules 2022, 12, 1672. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Jewell, C.M.; Bienstock, R.J.; Collins, J.B.; Cidlowski, J.A. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol. Cell. Biol. 2007, 27, 2266–2282. [Google Scholar] [CrossRef] [PubMed]
- Mac Namara, P.; Loughrey, H.C. Progesterone receptor A and B isoform expression in human osteoblasts. Calcif. Tissue Int. 1998, 63, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Tieszen, C.R.; Goyeneche, A.A.; Brandhagen, B.N.; Ortbahn, C.T.; Telleria, C.M. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer 2011, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—Mesenchymal and mesenchymal—Epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Drozd, A.M.; Mariani, L.; Guo, X.; Goitea, V.; Menezes, N.A.; Ferretti, E. Progesterone receptor modulates extraembryonic mesoderm and cardiac progenitor specification during mouse gastrulation. Int. J. Mol. Sci. 2022, 23, 10307. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Verhoog, N.J.D.; Van Looveren, K.; Dewaele, S.; Hochepied, T.; Eggermont, M.; Gilbert, B.; Munck, A.B.-D.; Vanderhaeghen, T.; Berghe, J.V.; et al. Point mutation i634a in the glucocorticoid receptor causes embryonic lethality by reduced ligand binding. J. Biol. Chem. 2022, 298, 101574. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Cidlowski, J.A. The physiology of human glucocorticoid receptor beta (hgrbeta) and glucocorticoid resistance. Ann. N. Y. Acad. Sci. 2006, 1069, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Witt, K.A.; Sandoval, K.E. Steroids and the blood-brain barrier: Therapeutic implications. Adv. Pharmacol. 2014, 71, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ihezie, S.A.; Mathew, I.E.; McBride, D.W.; Dienel, A.; Blackburn, S.L.; Pandit, P.K.T. Epigenetics in blood-brain barrier disruption. Fluids Barriers CNS 2021, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Forster, C.; Burek, M.; Romero, I.A.; Weksler, B.; Couraud, P.O.; Drenckhahn, D. Differential effects of hydrocortisone and tnfalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J. Physiol. 2008, 586, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Zhang, J. Systems wide analysis of CCM signaling complex alterations in CCM-deficient models using omics approaches. Methods Mol. Biol. 2020, 2152, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rigamonti, D.; Badr, A.; Zhang, J. CCM1 assures microvascular integrity during angiogenesis. Transl. Stroke Res. 2010, 1, 146–153. [Google Scholar] [CrossRef]
- Liu, H.; Rigamonti, D.; Badr, A.; Zhang, J. CCM1 regulates microvascular morphogenesis during angiogenesis. J. Vasc. Res. 2011, 48, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Flemming, K.D.; Lanzino, G. Are there differences in clinical presentation, radiologic findings, and outcomes in female patients with cavernous malformation? Acta Neurochir. 2023, 165, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Madsen, T.E.; Rexrode, K. Considering the use of female hormone therapy in women with cerebral cavernous malformations: An editorial. Neurology 2023, 100, 744–745. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, S.M.; Santos, A.N.; Flemming, K.D.; Schmidt, B.; Jabbarli, R.; Lanzino, G.; Sure, U.; Dammann, P. Female hormone therapy and risk of intracranial hemorrhage from cerebral cavernous malformations: A multicenter observational cohort study. Neurology 2023, 100, e1673–e1679. [Google Scholar] [CrossRef] [PubMed]
- Cici, M.; Dilmac, S.; Aytac, G.; Tanriover, G. Cerebral cavernous malformation proteins, CCM1, CCM2 and CCM3, are decreased in metastatic lesions in a murine breast carcinoma model. Biotech. Histochem. 2024, 99, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Cacciagli, P.; Sutera-Sardo, J.; Borges-Correia, A.; Roux, J.C.; Dorboz, I.; Desvignes, J.P.; Badens, C.; Delepine, M.; Lathrop, M.; Cau, P.; et al. Mutations in bcap31 cause a severe x-linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the golgi apparatus. Am. J. Hum. Genet. 2013, 93, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Jiang, X.; Padarti, A.; Goswami, D.G.; Smith, M.; Grajeda, B.; Bhalli, M.; Le, A.; Walker, W.E.; Zhang, J. Mpr-specific actions influence maintenance of the blood-brain barrier (BBB). Int. J. Mol. Sci. 2022, 23, 9684. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Qu, Y.; Gonzalez, E.M.; Smith, M.; Zhang, J. Emerging roles of CCM genes during tumorigenesis with potential application as novel biomarkers across major types of cancers. Oncol. Rep. 2020, 43, 1945–1963. [Google Scholar] [CrossRef] [PubMed]
- Vrinceanu, D.; Dumitru, M.; Marinescu, A.; Dorobat, B.; Palade, O.D.; Manole, F.; Muresian, H.; Popa-Cherecheanu, M.; Ciornei, C.M. New Insights into Cervicofacial Vascular Anomalies. J. Clin. Med. 2024, 13, 3515. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. ESM-SVM Models | B. CNNs Models | |
|---|---|---|
| F1 score | 0.948 | 0.934 |
| Specificity | 0.963 | 0.954 |
| Sensitivity | 0.958 | 0.947 |
| Accuracy | 0.961 | 0.950 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croft, J.; Grajeda, B.; Gao, L.; Abou-Fadel, J.; Badr, A.; Sheng, V.; Zhang, J. Whole-Genome Omics Elucidates the Role of CCM1 and Progesterone in Cerebral Cavernous Malformations within CmPn Networks. Diagnostics 2024, 14, 1895. https://doi.org/10.3390/diagnostics14171895
Croft J, Grajeda B, Gao L, Abou-Fadel J, Badr A, Sheng V, Zhang J. Whole-Genome Omics Elucidates the Role of CCM1 and Progesterone in Cerebral Cavernous Malformations within CmPn Networks. Diagnostics. 2024; 14(17):1895. https://doi.org/10.3390/diagnostics14171895
Chicago/Turabian StyleCroft, Jacob, Brian Grajeda, Liyuan Gao, Johnathan Abou-Fadel, Ahmed Badr, Victor Sheng, and Jun Zhang. 2024. "Whole-Genome Omics Elucidates the Role of CCM1 and Progesterone in Cerebral Cavernous Malformations within CmPn Networks" Diagnostics 14, no. 17: 1895. https://doi.org/10.3390/diagnostics14171895
APA StyleCroft, J., Grajeda, B., Gao, L., Abou-Fadel, J., Badr, A., Sheng, V., & Zhang, J. (2024). Whole-Genome Omics Elucidates the Role of CCM1 and Progesterone in Cerebral Cavernous Malformations within CmPn Networks. Diagnostics, 14(17), 1895. https://doi.org/10.3390/diagnostics14171895