
Assays for Monitoring Apixaban and Rivaroxaban in Emergency Settings, State-of-the-Art Routine Analysis, and Volumetric Absorptive Microsamples Deliver Discordant Results


 ,
,
Abstract
1. Introduction
2. Materials and Methods
- -
- methanol,
- -
- methanol-water 1:1 (vol/vol%),
- -
- methanol-acetonitrile 3:1 (vol/vol%), 1:1 (vol/vol%), or 1:3 (vol/vol%).
3. Results
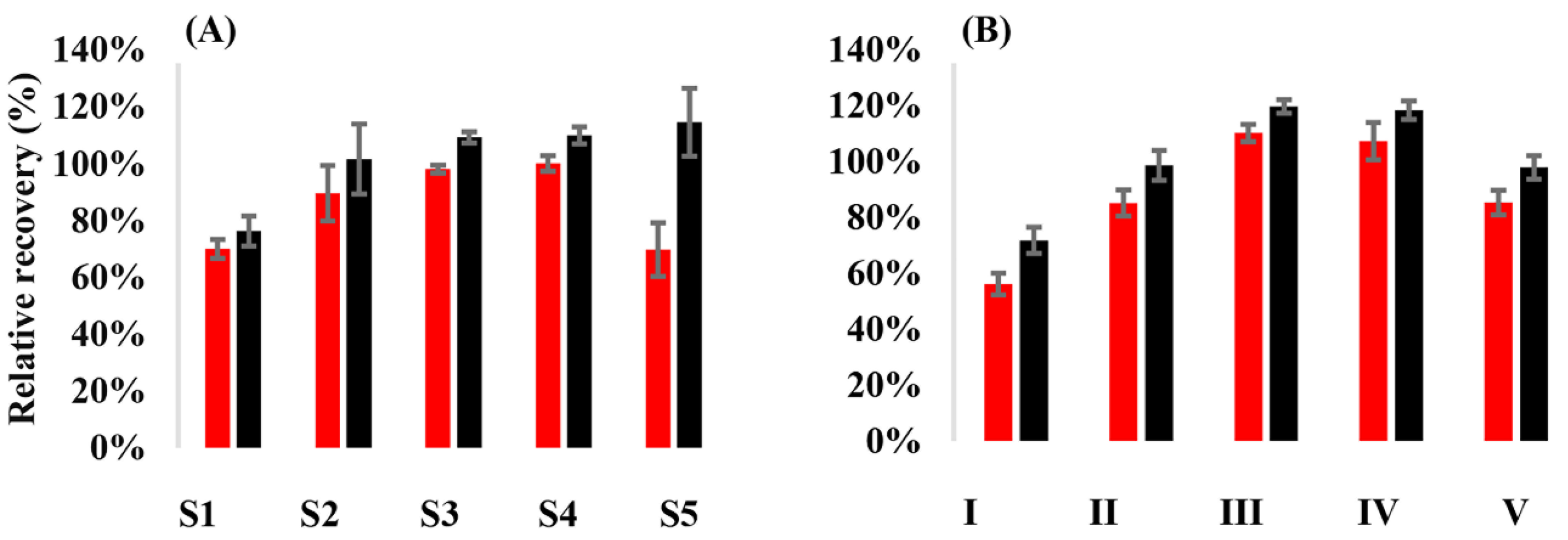
3.1. Relative Recovery of the Analytes from VAMS Samples
3.2. Technical Validation of the LC-MS/MS Assays
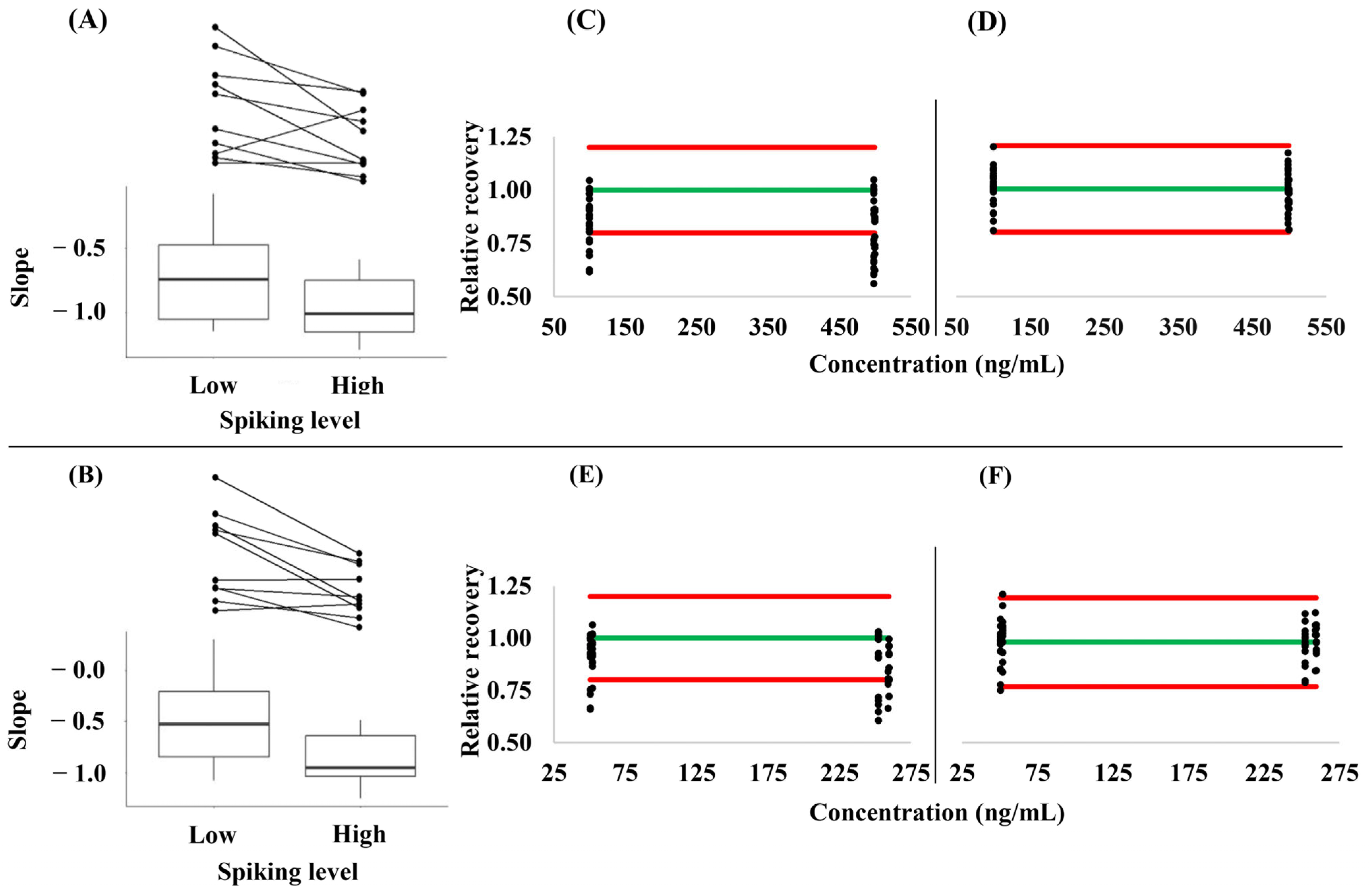
3.3. Hematocrit- and Concentration-Dependence of the Relative Analyte Recovery from VAMS Samples
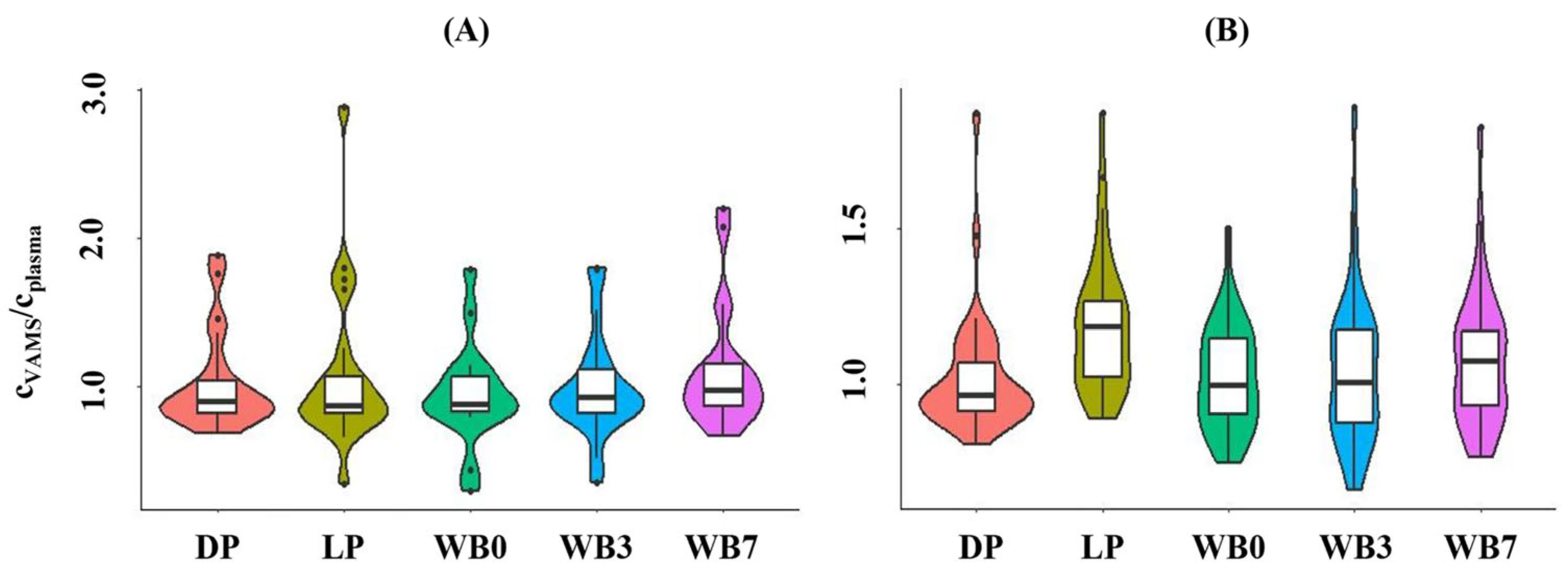
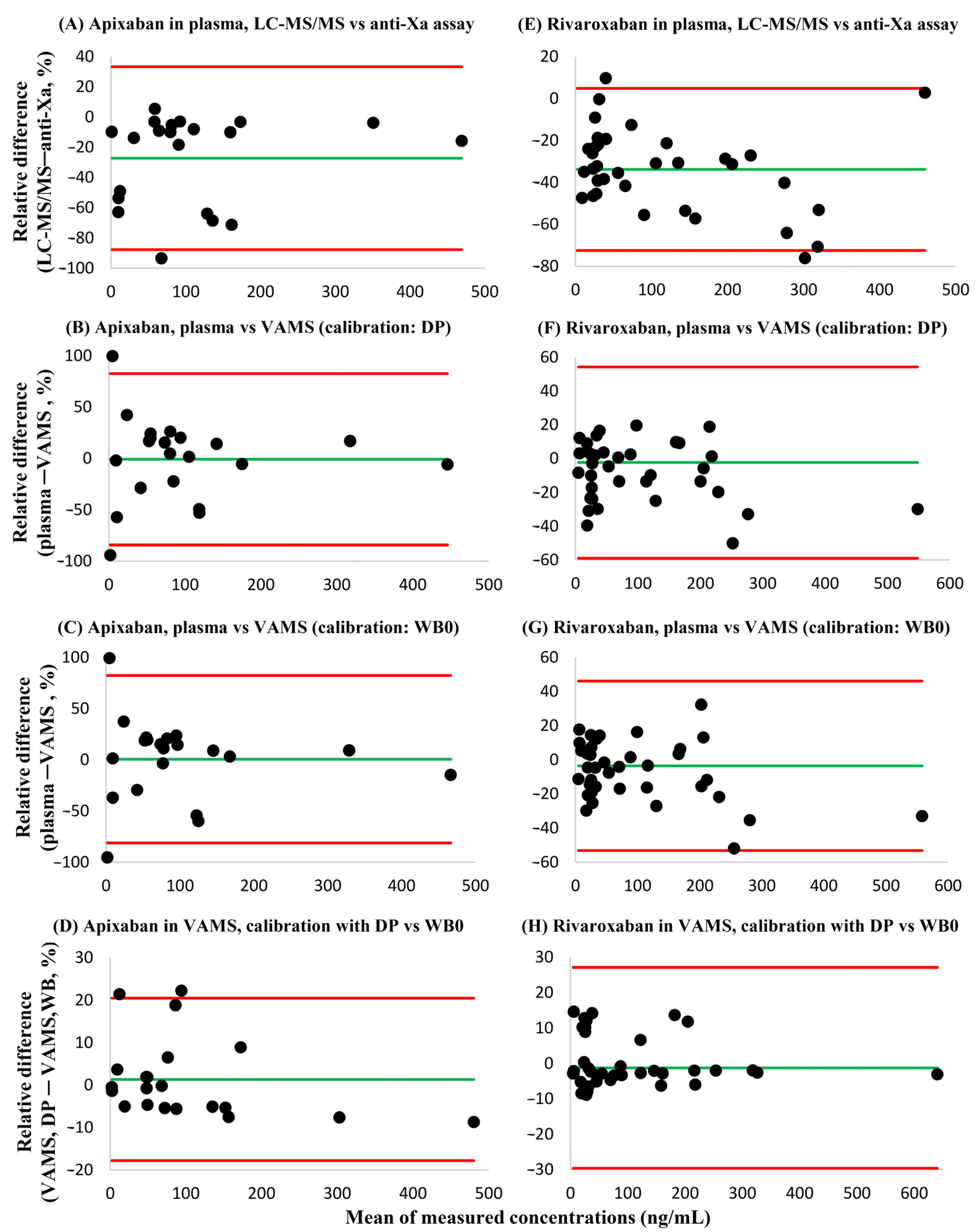
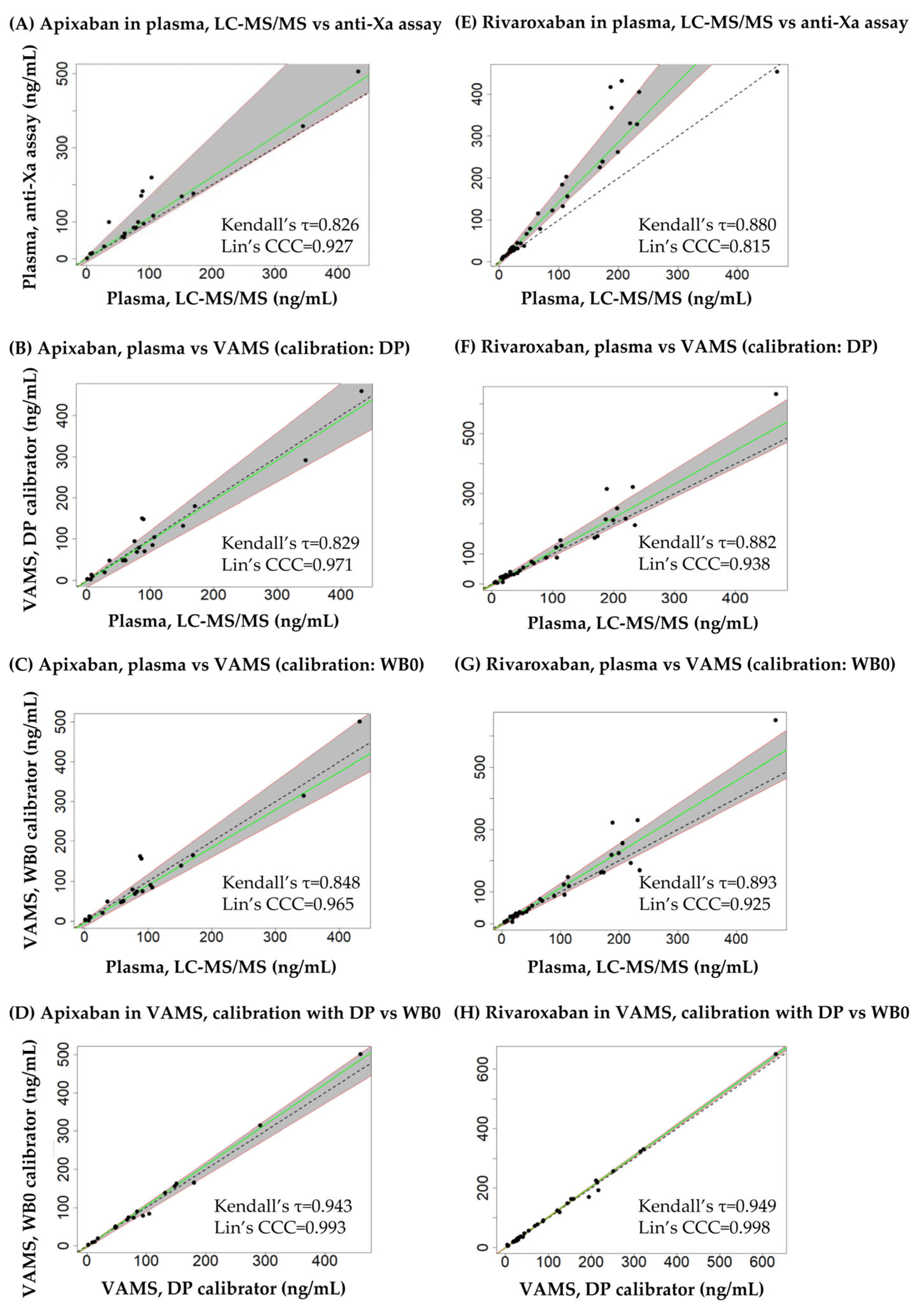
3.4. Cross-Validation Experiments Using Leftover Patient Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Wu, J.; Zhang, Y.; Liao, X.; Lei, Y. Anticoagulation therapy for non-valvular atrial fibrillation: A mini-review. Front. Med. 2020, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Stewart, H.M.; Russell, B.; Asiimwe, A.; Brobert, G. Time trends and treatment pathways in prescribing individual oral anticoagulants in patients with nonvalvular atrial fibrillation: An observational study of more than three million patients from Europe and the United States. Int. J. Clin. Pract. 2022, 2022, 6707985. [Google Scholar] [CrossRef]
- Chen, X.; Huang, W.; Sun, A.; Wang, L.; Mo, F.; Guo, W. Bleeding risks with novel oral anticoagulants especially RIV versus aspirin: A meta-analysis. Thromb. J. 2021, 19, 69. [Google Scholar] [CrossRef]
- McHorney, C.; Ashton, V.; Laliberté, F.; Germain, G.; Wynant, W.; Crivera, C.; Schein, J.R.; Lefebvre, P.; Peterson, E.D. Adherence to RIV compared with other oral anticoagulant agents among patients with nonvalvular atrial fibrillation. J. Manag. Care Spec. Pharm. 2017, 23, 980–988. [Google Scholar]
- Cools, F.; Johnson, D.; Camm, A.J.; Bassand, J.-P.; Verheugt, F.W.A.; Yang, S.; Tsiatis, A.; Fitzmaurice, D.A.; Goldhaber, S.Z.; Kayani, G.; et al. Risks associated with discontinuation of oral anticoagulation in newly diagnosed patients with atrial fibrillation: Results from the GARFIELD-AF Registry. J. Thromb. Haemost. 2021, 19, 2322–2334. [Google Scholar] [CrossRef]
- Moner-Banet, T.; Alberio, L.; Bart, P.-A. Does one dose really fit all? On the monitoring of direct oral anticoagulants: A review of the literature. Hämostaseologie 2020, 40, 184–200. [Google Scholar] [CrossRef]
- Rich, M.W. API monitoring. The Jury is still out. JACC Adv. 2022, 1, 100038. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Saunders, C.L.; Edwards, D.; Mant, J. Anticoagulation trends in adults aged 65 years and over with atrial fibrillation: A cohort study. Open Heart 2021, 8, e01737. [Google Scholar] [CrossRef]
- Camm, J.; Amarenco, P.; Haas, S.; Bach, M.; Kirchhof, P.; Kihls, S.; Lambelet, M.; Turpie, A.G.G.; XANTUS Investigators. Impact of gender: RIV for patients with atrail fibrillation in the XANTUS real-world prospective study. Clin. Cardiol. 2020, 43, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Shipkova, M. Pharmacokinetic and pharmacodynamic drug monitoring of direct-acting oral anticoagulants: Where do we stand? Ther. Drug Monit. 2019, 41, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Schroeder-Tanka, J.; Stooker, W.; Wissen, S.; Khorsand, N. Management of combined oral antithrombotic therapy by an antithrombotic stewardship program: A prospective study. Br. J. Clin. Pharmacol. 2022, 88, 4092–4099. [Google Scholar] [CrossRef]
- Dunois, C. Laboratory monitoring of direct-acting oral anticoagulants (DOACS). Biomedicines 2021, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Cafaro, A.; Conti, M.; Pigliasco, F.; Barco, S.; Bandettini, R.; Cangemi, G. Biological fluid microsampling for therapeutic drug monitoring: A narrative review. Biomedicines 2023, 11, 1962. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Bioanalytical Method Validation. EMEA/CHMP/192217/2009 Rev. 1 Corr. 2**. 21 July 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 3 July 2024).
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 3 July 2024).
- Passing, H.; Bablok, W. A new biometrical procedure for testing the equality of measurements from two different analytical methods. Application of linear regression procedures for method comparison studies in clinical chemistry, part I. J. Clin. Chem. Clin. Biochem. 1983, 21, 709–720. [Google Scholar] [CrossRef] [PubMed]
- McBRide, G.B. A Proposal for Strength-of-Agreement Criteria for Lin’s Concordance Correlation Coefficient. Prepared for the Ministry of Health; National Institute of Water & Atmospheric Research Ltd.: Hamilton, New Zealand, 2005; Available online: https://www.medcalc.org/download/pdf/McBride2005.pdf (accessed on 28 August 2024).
- Foerster, K.I.; Huppertz, A.; Meid, A.D.; Müller, O.J.; Rizos, T.; Tilemann, L.; Haefeli, W.E.; Burhenne, J. Dried-blood-spot technique to monitor direct oral anticoagulants: Clinical validation of a UPLC-MS/MS-based assay. Anal. Chem. 2018, 90, 9395–9402. [Google Scholar] [CrossRef]
- Jhang, R.-S.; Lin, S.-Y.; Peng, Y.-F.; Chao, H.-C.; Tsai, I.-L.; Lin, Y.-T.; Liao, H.-W.; Tang, S.-C.; Kuo, C.-H.; Jeng, J.-S. Using the PCI-IS method to simultaneously estimate blood volume and quantify nonvitamin K antagonists oral anticoagulant concentrations in dried blood spots. Anal. Chem. 2020, 92, 2511–2518. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Peng, Y.-F.; Huang, C.-F.; Kuo, C.-H.; Tang, S.-C.; Jeng, J.-S. Measurement of dabigatran concentration using finger prick dried blood spot sample collection. Front. Pharmacol. 2021, 12, 679431. [Google Scholar] [CrossRef]
- Iqbal, M.; Ullah, Z.; Ezzeldin, E.; Khalil, N.Y.; Al-Shakliah, N.S.; Alrasheed, A.; Ahmad, I.; Albehlal, L. A simple and sensitive HILIC-based UHPLC-MS/MS method for quantifying of RIV in dried blood spots: Application in comparison with the plasma sample method. J. Pharm. Biomed. Anal. 2021, 198, 114023. [Google Scholar] [CrossRef]
- Koster, R.A.; Alffenaar, J.-W.C.; Greijdanus, B.; Uges, D.R.A. Fast LC-MS/MS analysis of tacrolimus, sirolimus, everolimus and cyclosporin A in dried blood spots and the influence of the hematocrit and immunosuppressant concentration on recovery. Talanta 2013, 115, 47–54. [Google Scholar] [CrossRef]
- De Kesel, P.M.M.; Lambert, W.E.; Stove, C.P. Does volumetric absorptive microsampling eliminate the hematocrit bias for caffeine and paraxanthine in dried blood samples? A comparative study. Anal. Chim. Acta 2015, 881, 65–73. [Google Scholar] [CrossRef]
- Xie, I.; Xu, Y.; Anderson, M.; Wang, M.; Xue, L.; Breidinger, S.; Goykhman, D.; Woolf, E.J.; Bateman, K.P. Extractability-mediated stability bias and hematocrit impact: High extraction recovery is critical to feasibility of volumetric adsorptive microsampling (VAMS) in regulated bioanalysis. J. Pharm. Biomed. Anal. 2018, 156, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Aghai, F.; Schilling, B.; Kraus, S.; Grigoleit, G.U.; Kalogirou, C.; Goebeler, M.E.; Jung, P.; Pelzer, T.; Klinker, H.; et al. Volumetric absorptive microsampling (VAMS) for the quantification of ten kinase inhibitors and determination of their in vitro VAMS-to-plasma ratio. J. Pharm. Biomed. Anal. 2022, 211, 114623. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Aghai-Trommerschlaeger, F.; Kraus, S.; Grigoleit, G.U.; Gesierich, A.; Schilling, B.; Kalogirou, C.; Goebeler, M.-E.; Kurlbaum, M.; Klinker, H.; et al. Clinical validation and assessment of feasibility of volumetric absorptive microsampling (VAMS) for monitoring of nilotinib, cabozantinib, dabrafenib, trametinib, and ruxolitinib. J. Pharm. Biomed. Anal. 2023, 228, 115311. [Google Scholar] [CrossRef] [PubMed]
- Capiau, S.; Stove, C. Hematocrit prediction in volumetric absorptive microsamples. J. Pharm. Biomed. Anal. 2020, 190, 113491. [Google Scholar] [CrossRef]
- Jung, Y.; Yoo, S.; Kang, M.; Lim, H.; Lee, M.H.; Ryu, J.K.; Lee, J. Hematocrit determination using a volumetric absorptive microsampling technique in patients with pancreatic cancer. Korean J. Clin. Pharm. 2023, 33, 195–201. [Google Scholar] [CrossRef]
- Meihandoest, T.; Studt, J.-D.; Mendez, A.; Alberio, L.; Fontana, P.; Wuillemin, W.A.; Schmidt, A.; Graf, L.; Gerber, B.; Amstutz, U.; et al. Accuracy of a single, heparin-calibrated anti-Xa assay for the measurement of rivaroxaban, apixaban, and edoxaban drug concentrations: A prospective cross-sectional study. Front. Cardiovasc. Med. 2022, 9, 817826. [Google Scholar] [CrossRef]
- Kuhn, J.; Gripp, T.; Flieder, T.; Hammerschmidt, A.; Hendig, D.; Faust, I.; Knebbe, C.; Birschmann, I. Measurement of API, dabigatran, edoxaban and RIV in human plasma using automated solid-phase extraction combined with ultra-performance liquid chromatography-tandem mass spectrometry and its comparison with coagulation assays. Clin. Chim. Acta 2018, 486, 347–356. [Google Scholar] [CrossRef]
- Willekens, G.; Studt, J.-D.; Mendez, A.; Alberio, L.; Fontana, P.; Wuillemin, W.A.; Schmidt, A.; Graf, L.; Gerber, B.; Bovet, C.; et al. A universal anti-Xa assay for RIV, API, and edoxaban measurements: Method validation, diagnostic accuracy and external validation. Br. J. Haematol. 2021, 193, 1203–1212. [Google Scholar] [CrossRef]
- Derogis, P.B.M.; Sanches, L.R.; de Aranda, V.F.; Colombini, M.P.; Mangueira, C.L.P.; Katz, M.; Faulhaber, A.C.L.; Mendes, C.E.A.; Dos Santos Ferreira, C.E.; Franca, C.N.; et al. Determination of RIV in patient’s plasma samples by anti-Xa chromogenic test associated to high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS). PLoS ONE 2017, 12, e0171272. [Google Scholar] [CrossRef]
- Wasan, S.M.; Feland, N.; Grant, R.; Aston, C.E. Validation of API anti-factor Xa assay and impact of body weight. Thromb. Res. 2019, 182, 51–55. [Google Scholar] [CrossRef]
- Lessire, S.; Douxfils, J.; Pochet, L.; Sincq, A.-S.; Larock, A.-S.; Gourdin, M.; Dogné, J.-M.; Chtelain, B.; Mullier, F. Estimation of rivaroxaban plasma concentrations in the perioperative setting in patients with or without heparin bridging. Clin. Appl. Thromb. Hemost. 2018, 24, 129–138. [Google Scholar] [CrossRef] [PubMed]
 ). (●) shows the preparation of dried whole blood calibrators, (●) marks the preparation of dried plasma calibrators, and (●) displays the application of liquid plasma calibrators.
). (●) shows the preparation of dried whole blood calibrators, (●) marks the preparation of dried plasma calibrators, and (●) displays the application of liquid plasma calibrators.
). (●) shows the preparation of dried whole blood calibrators, (●) marks the preparation of dried plasma calibrators, and (●) displays the application of liquid plasma calibrators.
). (●) shows the preparation of dried whole blood calibrators, (●) marks the preparation of dried plasma calibrators, and (●) displays the application of liquid plasma calibrators.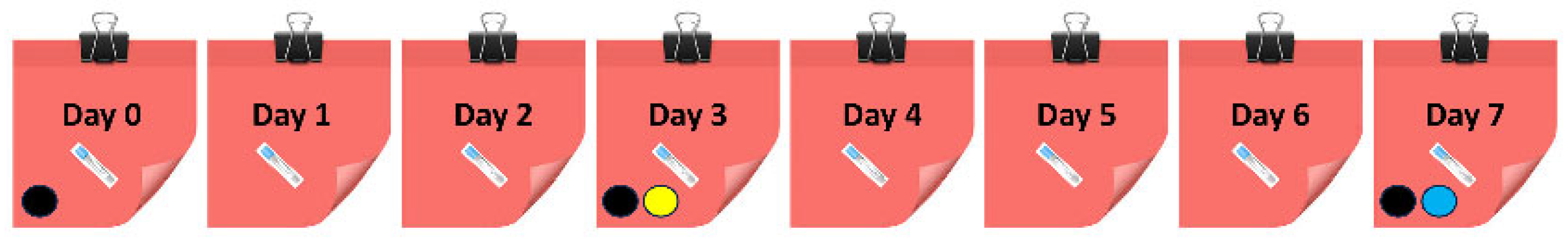

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance Characteristics | Within-Run Experiment | Between-Run Experiment, Day 1 | Between-Run Experiment, Day 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QC 1 | QC 2 | QC 3 | QC 4 | QC 5 | QC 6 | QC 2 | QC 3 | QC 5 | QC 2 | QC 3 | QC 5 | |
| Apixaban in plasma | ||||||||||||
| Accuracy (%) | 99.9 | 105.3 | 95.6 | 103.5 | 114.0 | 111.3 | 102.1 | 98.1 | 104.4 | 97.2 | 94.4 | 104.1 |
| RSD (%) | 4.9 | 4.3 | 12.8 | 6.8 | 4.1 | 3.9 | 2.8 | 5.2 | 5.4 | 5.1 | 6.2 | 5.6 |
| Apixaban in VAMS, whole blood calibrators | ||||||||||||
| Accuracy (%) | 94.1 | 94.7 | 93.8 | 91.7 | 95.8 | 90.6 | 99.3 | 95.9 | 96.9 | 97.0 | 94.0 | 98.3 |
| RSD (%) | 8.0 | 6.5 | 4.8 | 7.8 | 3.4 | 7.1 | 4.8 | 5.3 | 2.6 | 5.1 | 6.3 | 4.7 |
| Apixaban in VAMS, dried plasma calibrators | ||||||||||||
| Accuracy (%) | 100.0 | 98.2 | 96.9 | 94.6 | 98.8 | 93.4 | 102.8 | 99.4 | 100.5 | 102.3 | 98.6 | 102.6 |
| RSD (%) | 7.7 | 6.5 | 4.8 | 7.8 | 3.4 | 7.1 | 4.8 | 5.3 | 2.6 | 5.0 | 6.3 | 4.7 |
| Apixaban in VAMS, liquid plasma calibrators | ||||||||||||
| Accuracy (%) | 105.4 | 114.5 | 114.3 | 112.4 | 117.7 | 111.4 | 115.8 | 113.0 | 115.2 | 120.6 | 116.6 | 121.7 |
| RSD (%) | 8.7 | 6.6 | 4.8 | 7.8 | 3.4 | 7.1 | 4.9 | 5.4 | 2.6 | 5.1 | 6.3 | 4.7 |
| Rivaroxaban in plasma | ||||||||||||
| Accuracy (%) | 98.1 | 103.8 | 107.8 | 105.7 | 111.7 | 107.0 | 101.2 | 101.2 | 104.4 | 100.5 | 100.1 | 100.8 |
| RSD (%) | 3.8 | 4.3 | 2.2 | 1.9 | 2.4 | 1.7 | 6.9 | 3.3 | 5.7 | 4.5 | 3.3 | 1.7 |
| Rivaroxaban in VAMS, whole blood calibrators | ||||||||||||
| Accuracy (%) | 100.7 | 96.6 | 97.7 | 93.3 | 97.1 | 92.3 | 97.8 | 96.1 | 97.2 | 94.8 | 93.6 | 98.5 |
| RSD (%) | 10.9 | 4.7 | 4.6 | 5.6 | 2.9 | 6.1 | 4.7 | 3.6 | 1.4 | 8.1 | 5.9 | 3.5 |
| Rivaroxaban in VAMS, dried plasma calibrators | ||||||||||||
| Accuracy (%) | 100.2 | 96.5 | 97.7 | 93.3 | 97.2 | 92.3 | 97.8 | 96.2 | 97.3 | 94.5 | 92.4 | 96.4 |
| RSD (%) | 11.0 | 4.7 | 4.6 | 5.6 | 2.9 | 6.1 | 4.7 | 3.6 | 1.4 | 7.9 | 5.8 | 3.5 |
| Rivaroxaban in VAMS, liquid plasma calibrators | ||||||||||||
| Accuracy (%) | 116.5 | 121.3 | 124.0 | 119.1 | 124.3 | 118.2 | 117.5 | 117.4 | 120.4 | 126.2 | 122.1 | 126.2 |
| RSD (%) | 12.1 | 4.7 | 4.6 | 5.7 | 2.9 | 6.1 | 4.9 | 3.6 | 1.4 | 7.8 | 5.7 | 3.5 |
| Matrix | API | RIV | ||
|---|---|---|---|---|
| Low Spiking Level | High Spiking Level | Low Spiking Level | High Spiking Level | |
| Plasma | 0.86 ± 0.05 (5.4%) | 1.01 ± 0.07 (7.2%) | 0.90 ± 0.13 (15.0%) | 1.05 ± 0.06 (6.1%) |
| Dried whole blood | 1.00 ± 0.04 (3.6%) | 0.96 ± 0.05 (5.5%) | 1.03 ± 0.05 (4.9%) | 0.94 ± 0.06 (6.7%) |
| Analyte | Spiking Level | Spiked Concentration (ng/mL) | Relative Recovery (%) | |
|---|---|---|---|---|
| Plasma | VAMS Samples | |||
| API | Low | 53.2 | 94.3–118 | |
| 49.8 | 98.0–110 | |||
| Middle | 107 | 93.4–113 | ||
| 99.6 | 91.3–105 | |||
| High | 428 | 89.5–103 | ||
| 498 | 96.3–105 | |||
| RIV | Low | 53.2 | 96.1–106 | |
| 25.2 | 94.0–106 | |||
| Middle | 107 | 102–109 | ||
| 50.4 | 90.1–100 | |||
| High | 428 | 100–107 | ||
| 252 | 95.1–99.2 | |||
| Substance | Approaches Compared | Slope (95% CI) | Intercept (95% CI) | h Statistic |
|---|---|---|---|---|
| Apixaban | Plasma, LC-MS/MS and plasma, Anti-Xa | 1.11 (1.03–1.59) | 0.12 (−12.7–5.73) | 0.91 |
| Plasma, LC-MS/MS and VAMS (calibrator: DP) | 0.98 (0.86–1.23) | −2.26 (−18.3–1.74) | 0.91 | |
| Plasma, LC-MS/MS and VAMS (calibrator: WB0) | 0.95 (0.87–1.16) | −4.25 (−13.6–1.80) | 1.21 | |
| VAMS, DP calibrator and WB0 calibrator | 1.06 (0.94–1.09) | −0.91 (−4.92–0.23) | 0.91 | |
| Rivaroxaban | Plasma, LC-MS/MS and plasma, Anti-Xa | 1.43 (1.34–1.75) | −1.14 (−8.39–0.85) | 0.89 |
| Plasma, LC-MS/MS and VAMS (calibrator: DP) | 1.12 (0.97–1.26) | −2.27 (−7.20–1.76) | 0.89 | |
| Plasma, LC-MS/MS and VAMS (calibrator: WB0) | 1.15 (0.97–1.27) | −2.09 (−6.85–1.40) | 1.12 | |
| VAMS, DP calibrator and WB0 calibrator | 1.02 (1.00–1.03) | −0.05 (−1.21–0.87) | 0.89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehér, A.; Vincze, I.; Rudge, J.; Domján, G.; Vásárhelyi, B.; Karvaly, G.B. Assays for Monitoring Apixaban and Rivaroxaban in Emergency Settings, State-of-the-Art Routine Analysis, and Volumetric Absorptive Microsamples Deliver Discordant Results. Diagnostics 2024, 14, 1939. https://doi.org/10.3390/diagnostics14171939
Fehér A, Vincze I, Rudge J, Domján G, Vásárhelyi B, Karvaly GB. Assays for Monitoring Apixaban and Rivaroxaban in Emergency Settings, State-of-the-Art Routine Analysis, and Volumetric Absorptive Microsamples Deliver Discordant Results. Diagnostics. 2024; 14(17):1939. https://doi.org/10.3390/diagnostics14171939
Chicago/Turabian StyleFehér, Adrienne, István Vincze, James Rudge, Gyula Domján, Barna Vásárhelyi, and Gellért Balázs Karvaly. 2024. "Assays for Monitoring Apixaban and Rivaroxaban in Emergency Settings, State-of-the-Art Routine Analysis, and Volumetric Absorptive Microsamples Deliver Discordant Results" Diagnostics 14, no. 17: 1939. https://doi.org/10.3390/diagnostics14171939
APA StyleFehér, A., Vincze, I., Rudge, J., Domján, G., Vásárhelyi, B., & Karvaly, G. B. (2024). Assays for Monitoring Apixaban and Rivaroxaban in Emergency Settings, State-of-the-Art Routine Analysis, and Volumetric Absorptive Microsamples Deliver Discordant Results. Diagnostics, 14(17), 1939. https://doi.org/10.3390/diagnostics14171939