
Body Height of MPS I and II Patients after Hematopoietic Stem Cell Transplantation: The Impact of Dermatan Sulphate

 , , , and
, , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Anthropometric Measurements
2.2. GAG Electrophoresis
3. Patient Characteristics
3.1. Overall Characteristics
3.2. Clinical Outcome
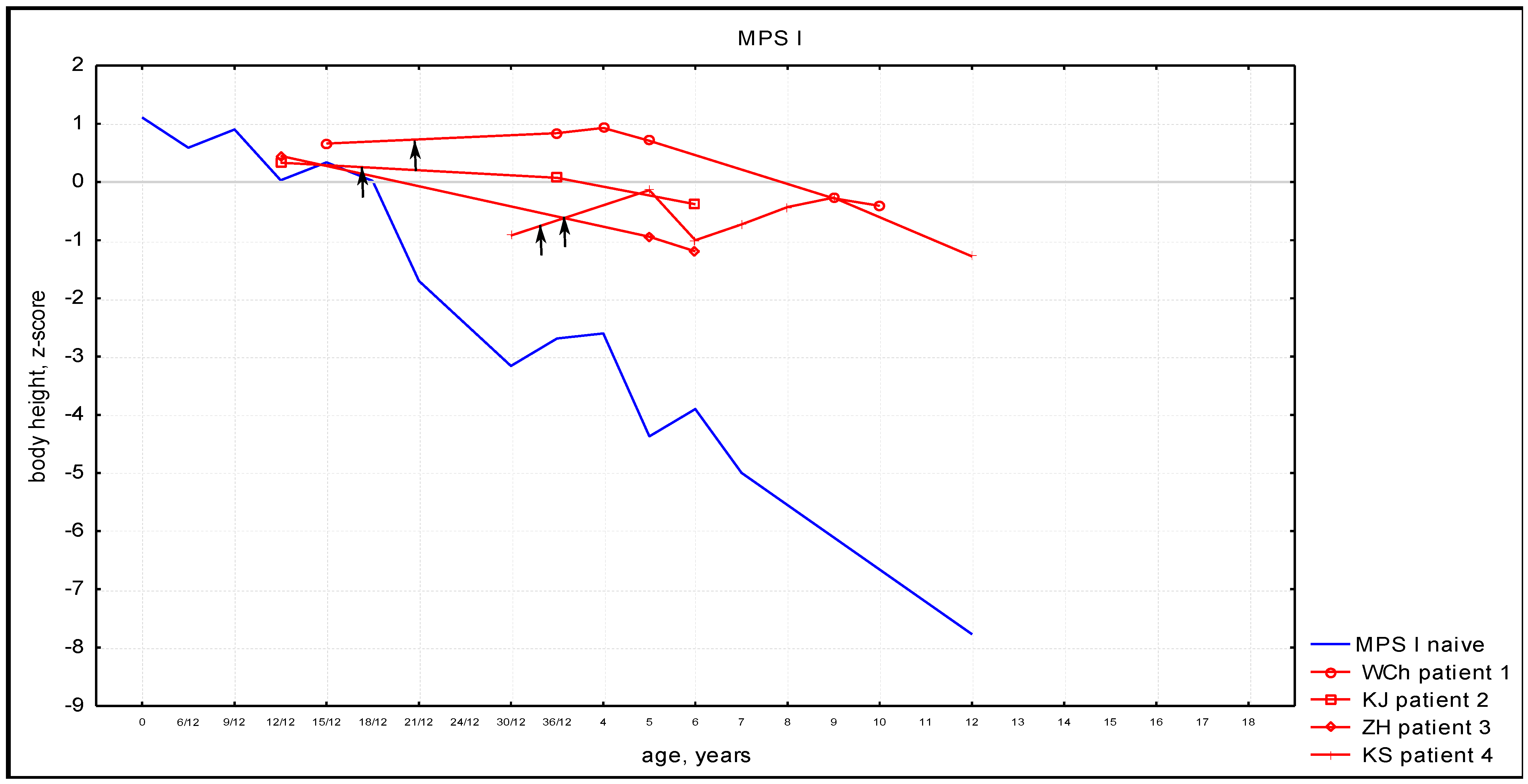
3.3. Anthropometric Phenotype
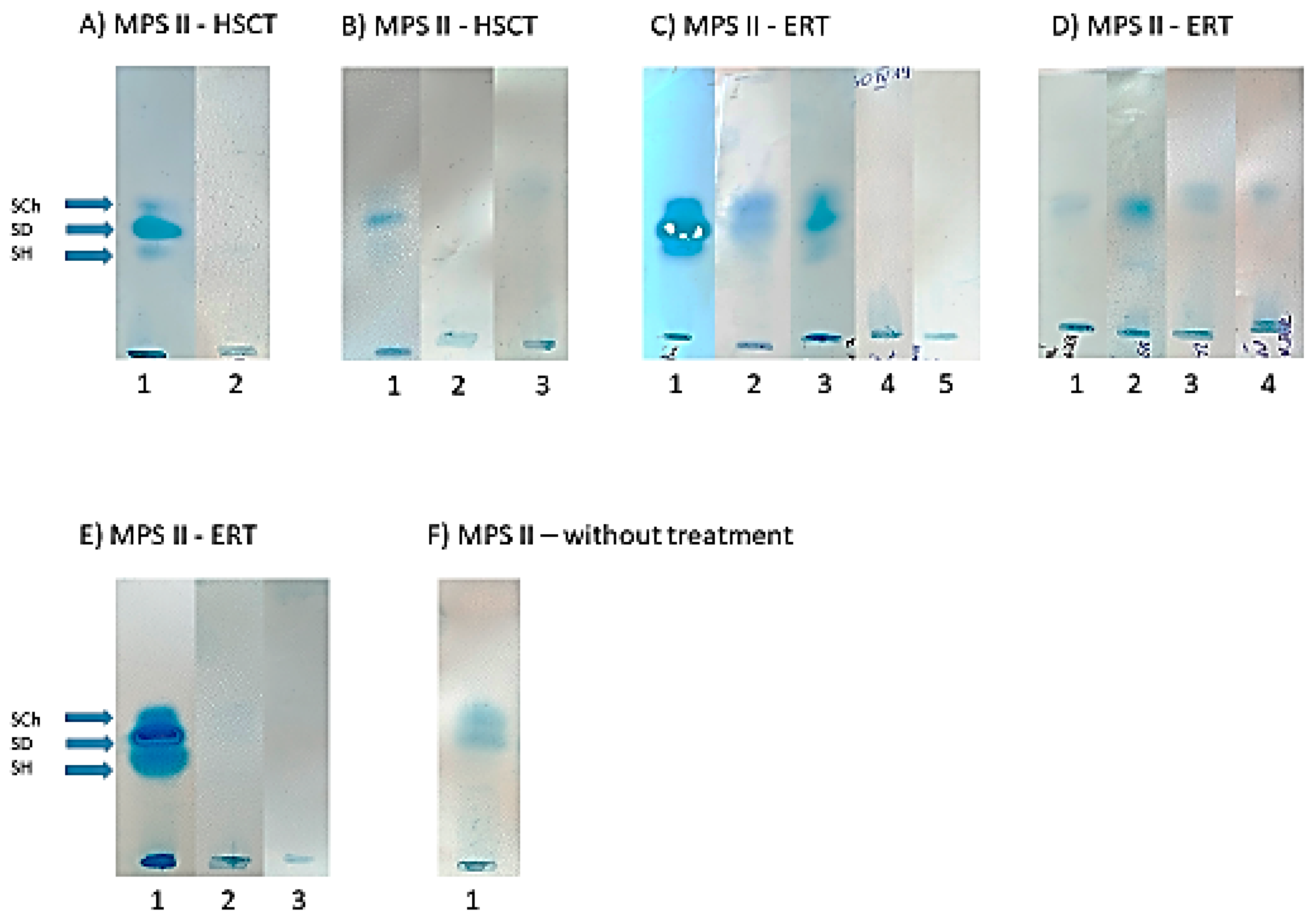
3.4. Urinary GAG Electrophoresis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiśniewska, K.; Wolski, J.; Gaffke, L.; Cyske, Z.; Pierzynowska, K.; Węgrzyn, G. Misdiagnosis in mucopolysaccharidoses. J. Appl. Genet. 2022, 63, 475–495. [Google Scholar] [CrossRef]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef]
- Ago, Y.; Rintz, E.; Musini, K.S.; Ma, Z.; Tomatsu, S. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int. J. Mol. Sci. 2024, 25, 1113. [Google Scholar] [CrossRef]
- Kubaski, F.; De Poswar, F.D.; Michelin-Tirelli, K.; Burin, M.G.; Rojas-Málaga, D.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Giugliani, R. Diagnosis of Mucopolysaccharidoses. Diagnostics 2020, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Kubaski, F.; De Poswar, F.D.; Michelin-Tirelli, K.; Da Matte, U.D.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagnostics 2020, 10, 161. [Google Scholar] [CrossRef]
- D’avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21, 1258. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Beck, M. Mucopolysaccharidosis Type II (Hunter Syndrome): Clinical picture and treatment. Curr. Pharm. Biotechnol. 2011, 12, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, J.R.; Hugh-Jones, K.; Barrett, A.J.; Byrom, N.; Chambers, D.; Henry, K.; James, D.C.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 318, 709–712. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Transplant. Cell. Ther. 2019, 25, e226–e246. [Google Scholar] [CrossRef]
- Hopwood, J.J.; Harrison, J.R. High-resolution electrophoresis of urinary glycosamino glycans: An improved screening test for the mucopolysaccharidoses. Anal. Biochem. 1982, 119, 120–127. [Google Scholar] [CrossRef]
- Różdżyńska-Świątkowska, A.; Jurecka, A.; Cieślik, J.; Tylki-Szymańska, A. Growth patterns in children with mucopolysaccharidosis I and II. World J. Pediatr. 2014, 11, 226–231. [Google Scholar] [CrossRef]
- Tylki-Szymanska, A.; Rozdzynska, A.; Jurecka, A.; Marucha, J.; Czartoryska, B. Anthropometric data of 14 patients with mucopolysaccharidosis I: Retrospective analysis and efficacy of recombinant human α-l-iduronidase (laronidase). Mol. Genet. Metab. 2009, 99, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Różdżyńska-Świątkowska, A.; Zielińska, A.; Tylki-Szymańska, A. Comparison of growth dynamics in different types of MPS: An attempt to explain the causes. Orphanet J. Rare Dis. 2022, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Żuber, Z.; Różdżyńska-Świątkowska, A.; Jurecka, A.; Tylki-Szymańska, A. The effect of recombinant human iduronate-2-sulfatase (Idursulfase) on growth in young patients with mucopolysaccharidosis type II. PLoS ONE 2014, 9, e85074. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Parini, R.; Harmatz, P.; Giugliani, R.; Fang, J.; Mendelsohn, N.J. The effect of idursulfase on growth in patients with Hunter syndrome: Data from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2013, 109, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cattoni, A.; Chiaraluce, S.; Gasperini, S.; Molinari, S.; Biondi, A.; Rovelli, A.; Parini, R. Growth patterns in children with mucopolysaccharidosis type I-Hurler after hematopoietic stem cell transplantation: Comparison with untreated patients. Mol. Genet. Metab. Rep. 2021, 28, 100787. [Google Scholar] [CrossRef]
- Guffon, N.; Pettazzoni, M.; Pangaud, N.; Garin, C.; Lina-Granade, G.; Plault, C.; Mottolese, C.; Froissart, R.; Fouilhoux, A. Long term disease burden post-transplantation: Three decades of observations in 25 Hurler patients successfully treated with hematopoietic stem cell transplantation (HSCT). Orphanet J. Rare Dis. 2021, 16, 1–20. [Google Scholar] [CrossRef]
- Patel, P.; Suzuki, Y.; Tanaka, A.; Yabe, H.; Kato, S.; Shimada, T.; Mason, R.W.; Orii, K.E.; Fukao, T.; Orii, T.; et al. Impact of Enzyme Replacement Therapy and Hematopoietic Stem Cell Therapy on Growth in Patients with Hunter Syndrome. Mol. Genet. Metab. Rep. 2014, 1, 184–196. [Google Scholar] [CrossRef]
- Santi, L.; De Ponti, G.; Dina, G.; Pievani, A.; Corsi, A.; Riminucci, M.; Khan, S.; Sawamoto, K.; Antolini, L.; Gregori, S.; et al. Neonatal combination therapy improves some of the clinical manifestations in the Mucopolysaccharidosis type I murine model. Mol. Genet. Metab. 2020, 130, 197–208. [Google Scholar] [CrossRef]
- Pievani, A.; Azario, I.M.R.; Antolini, L.; Shimada, T.; Patel, P.; Remoli, C.; Rambaldi, B.; Valsecchi, M.G.; Riminucci, M.; Biondi, A.; et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood 2015, 125, 1662–1671. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and bone disease in mucopolysaccharidoses VI and VII: Identification of new therapeutic targets and biomarkers using animal models. Pediatr. Res. 2005, 57, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Hinek, A.; Wilson, S.E. Impaired elastogenesis in Hurler disease—Dermatan sulfate accumulation linked to deficiency in elastin-binding protein and elastic fiber assembly. Am. J. Pathol. 2000, 156, 925–938. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. Type of MPS Genotype | Age | Anthropometric Analyses | Alpha-Iduronidase (MPS I) or Iduronate 2-Sulfatase (MPS II) Activity in DBS | Alpha-Iduronidase (MPS I) or Iduronate 2-Sulfatase (MPS II) Activity in Leukocytes | GAGs in Urine—Quantitative Analysis with DMB | GAGs in Urine—Semi-Quantitative Analysis with Cetylpyridinium Chloride | GAGs Electrophoresis | Clinical Outcome |
|---|---|---|---|---|---|---|---|---|
| 1 MPS I c.208C > T, p.(Gln70*)/ c.208C > T, p.(Gln70*) | 13 m | Weight 10.9 kg (50 pc) Height 79.5 cm (50 pc) | <1.1 (limit of quantification) µmol/L/h (ref. > 3.0 µmol/L/h) | 10 nmol/mg protein/18 h (129 ± 40.5) | 158.7 (4.21–18.7) mg/mmol creatinine) | 699 (151 ± 77) mg/g creatinine | DS and subtle HS | inguinal hernia, subtle corneal clouding, thickened mitral valve, mild liver enlargement, limited range of motion in the glenohumeral joints |
| 5.5 y (3 y after HSCT) | Weight 21.2 kg (50 pc) Height 113.8 cm (35 pc) | 1.7 µmol/L/h (ref. > 3.0 µmol/L/h) | 180 nmol/mg protein/18 h (129 ± 40.5) | 17.6 (4.21–18.7 mg/mmol creatinine) | 55 (113 ± 46) mg/g creatinine | Normal results | stable thickening of mitral valve, corneal opacity over the entire surface, qualification for adenotomy, normal cognitive development, stable limitation of the range of motion in the glenohumeral joints, narrowing of the spinal canal (5.5 y) | |
| 2 MPS I c.208C > T, p.(Gln70*)/ c.208C > T, p.(Gln70*) | 14 m | Weight 11.6 kg (50–75 pc) Height 80.5 cm (50–75 pc) | <1.1 (limit of quantification) µmol/L/h (ref. > 3.0 µmol/L/h) | 9.72 nmol/mg protein/18 h (129 ± 40.5) | 137 (4.21–18.7) mg/mmol creatinine) | 648 (151 ± 77) mg/g creatinine | DS and subtle HS | bilateral hearing loss, thickened mitral valve, mild liver enlargement, limited range of motion in the glenohumeral joints |
| 5.5 y | Weight 20.3 kg (19 pc) Height 109.2 cm (12 pc) | 5.9 µmol/L/h (ref. > 3.0 µmol/L/h) | 178 nmol/mg protein/18 h (129 ± 40.5) | 16.4 (4.21–18.7 mg/mmol creatinine) | 55 (113 ± 46) mg/g creatinine | Traces of DS and HS | mild hearing loss, carpal tunnel syndrome diagnosed at 5 y, normal cognitive development, stable limitation of the range of motion in the glenohumeral joints, carpal tunnel syndromne (5 y) | |
| 3 MPS I c.208C > T, p.(Gln70*)/ c.208C > T, p.(Gln70*) | 13 m | Weight 10.3 kg (10–25 pc) Height 80.6 cm (75–90 pc) | <1.1 (limit of quantification) µmol/L/h (ref. > 3.0 µmol/L/h) | 0.01 nmol/mg protein/18 h (129 ± 40.5) | 253 (4.21–18.7) mg/mmol creatinine) | 1603 (151 ± 77) mg/g creatinine | DS | Thickened mitral valve, bilateral hearing loss, corneal clouding, mild liver enlargement, limited range of motion in the glenohumeral joints |
| 12 y | Weight 40.2 kg (77 pc) Height 137.2 cm (34 pc) | 3.9 µmol/L/h (ref. > 3.0 µmol/L/h) | 87 nmol/mg protein/18 h (129 ± 40.5) | 15.3 (4.21–18.7 mg/mmol creatinine) | 66 (115 ± 61) mg/g creatinine | Normal results | no carpal tunnel syndrome, progressive thoracic kyphosis, bilateral hip dysplasia, normal intellectual development, stable thickening of mitral valve | |
| 4 MPS I c.208C > T, p.(Gln70*)/ c.208C > T, p.(Gln70*) | 12 y (age at diagnosis: 16 months) | Weight 49.7 kg (78 pc) Height 144.3 cm (10 pc) | 4.1 µmol/L/h (ref. > 3.0 µmol/L/h) | 132 nmol/mg protein/18 h (129 ± 40.5) | 9.6 (4.21–18.7 mg/mmol creatinine) | 134 (115 ± 61) mg/g creatinine | Normal results | Bilateral hip dysplasia, progressive lumbar lordosis, mild hearing loss, corneal opacity over the entire surface, carpal tunnel syndrome (12 y), normal intellectual development |
| 5 MPS II c.1134_1152dup, p.(Asp385ProfsX7) | 14 m | Weight 15.3 kg (90–97 pc) Height 86.5 cm (>97 pc) | <0.8 (limit of detection) µmol/L/h (ref. > 5.6 µmol/L/h) | 2.8 nmol/mg protein/4 h (354 ± 85.9) | 111.7 (9.52–26.9 mg/mmol creatinine) | 944 (151 ± 77) mg/g creatinine | DS, HS | limited range of motion in the glenohumeral joints, qualified for adenectomy, mild liver enlargement, thickened mitral valve |
| 5 y (3 y after HSCT) | Weight 20.1 kg (62 pc) Height 116.2 cm (91 pc) | 3.1 µmol/L/h (ref. > 5.6 µmol/L/h) | 28.8 nmol/mg protein/4 h (354 ± 85.9) | 13.2 (9.52–26.9 mg/mmol creatinine) | 207 (151 ± 77) mg/g creatinine | Normal results | normal intellectual development, stable limitation of the range of motion in the glenohumeral joints, stable thickening of the mitral valve | |
| 6 MPS II IDS, loss of exons 01 to 08 | 14 m | Weight 15.3 kg (90–97 pc) Height 86.5 cm (>97 pc) | <2.8 (limit of quantification) µmol/L/h (ref. > 5.6 µmol/L/h) | 2.8 nmol/mg protein/4 h (354 ± 85.9) | 103.7 (9.52–26.9 mg/mmol creatinine) | 957 (151 ± 77) mg/g creatinine | DS, HS | moderate hearing loss, qualified for adenectomy, unilateral hip dysplasia, limited range of motion in the glenohumeral and hip joints, mild liver enlargement, thickened mitral valve |
| 3 y 2 m (1 y and 3 m after HSCT) | Weight 22.3 kg (>97 pc) Height 102.8 cm (92 pc) | 4.2 µmol/L/h (ref. > 3.0 µmol/L/h) | 35.9 nmol/mg protein/4 h (354 ± 85.9) | 13.2 (9.52–26.9 mg/mmol creatinine) | 226 (151 ± 77) mg/g creatinine | Normal results | moderate hearing loss, hearing aids, stable limitation of the range of motion in the glenohumeral and hip joints, stable thickening of mitral valve |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipiński, P.; Różdżyńska-Świątkowska, A.; Ługowska, A.; Marucha, J.; Drabko, K.; Tylki-Szymańska, A. Body Height of MPS I and II Patients after Hematopoietic Stem Cell Transplantation: The Impact of Dermatan Sulphate. Diagnostics 2024, 14, 1956. https://doi.org/10.3390/diagnostics14171956
Lipiński P, Różdżyńska-Świątkowska A, Ługowska A, Marucha J, Drabko K, Tylki-Szymańska A. Body Height of MPS I and II Patients after Hematopoietic Stem Cell Transplantation: The Impact of Dermatan Sulphate. Diagnostics. 2024; 14(17):1956. https://doi.org/10.3390/diagnostics14171956
Chicago/Turabian StyleLipiński, Patryk, Agnieszka Różdżyńska-Świątkowska, Agnieszka Ługowska, Jolanta Marucha, Katarzyna Drabko, and Anna Tylki-Szymańska. 2024. "Body Height of MPS I and II Patients after Hematopoietic Stem Cell Transplantation: The Impact of Dermatan Sulphate" Diagnostics 14, no. 17: 1956. https://doi.org/10.3390/diagnostics14171956