
Different Shades of Desmoid-Type Fibromatosis (DTF): Detection of Noval Mutations in the Clinicopathologic Analysis of 32 Cases


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
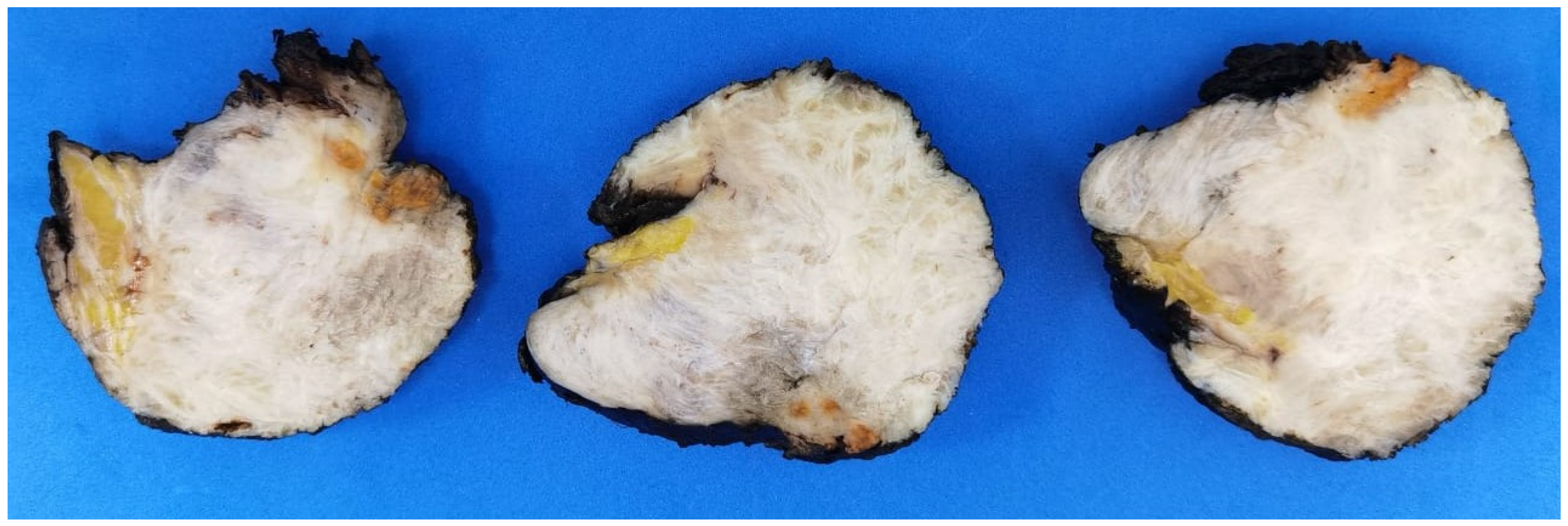
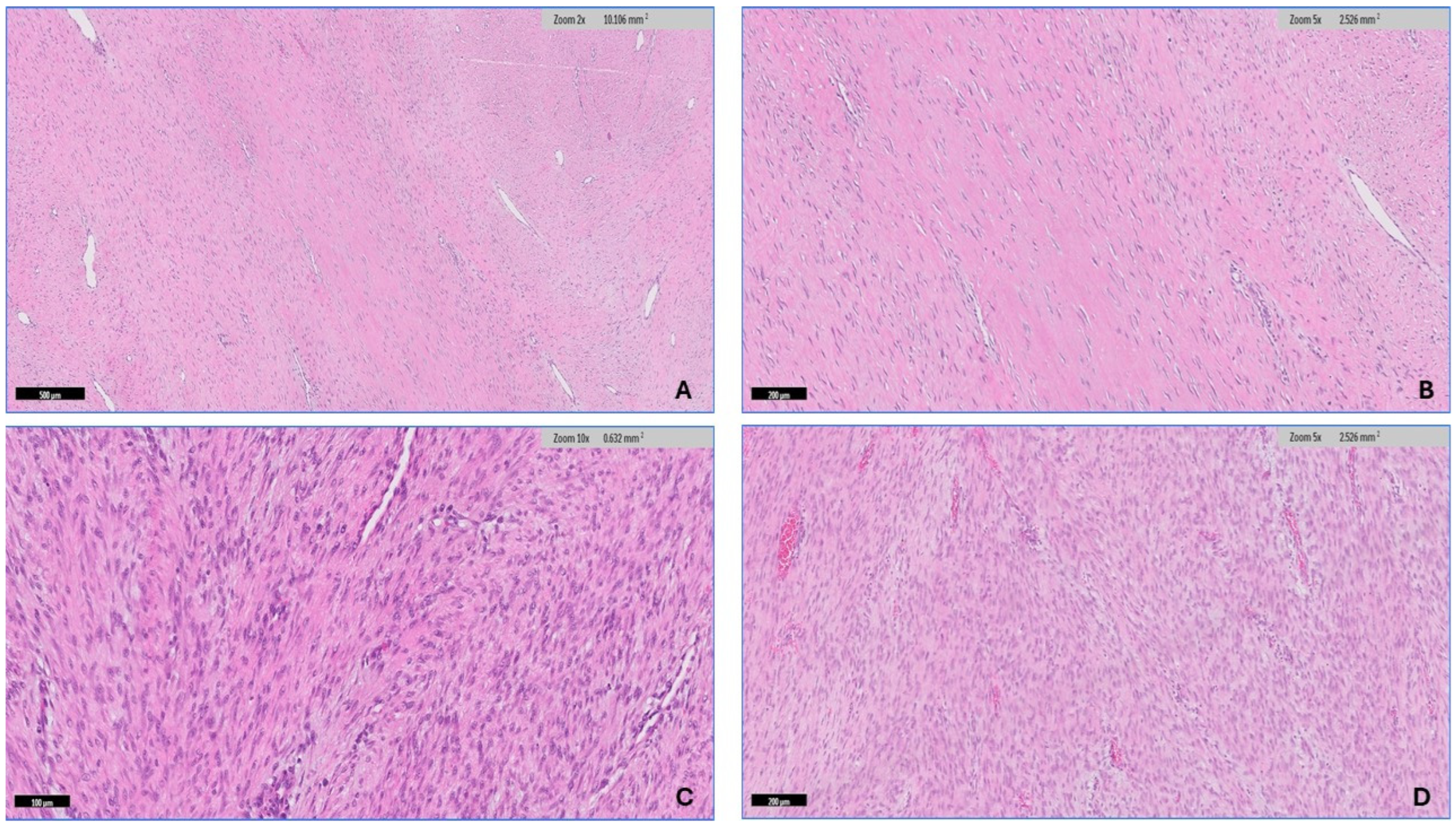
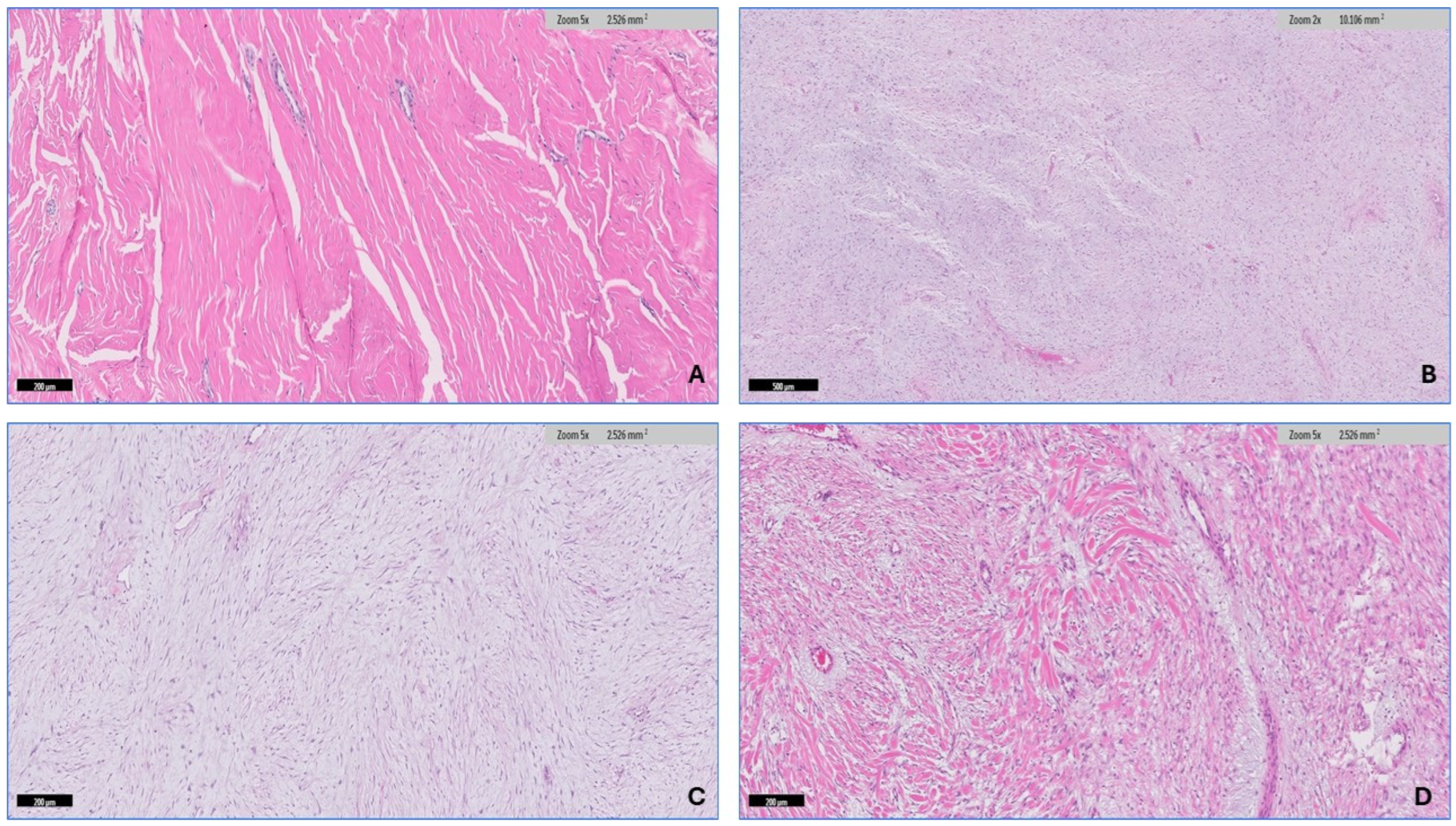
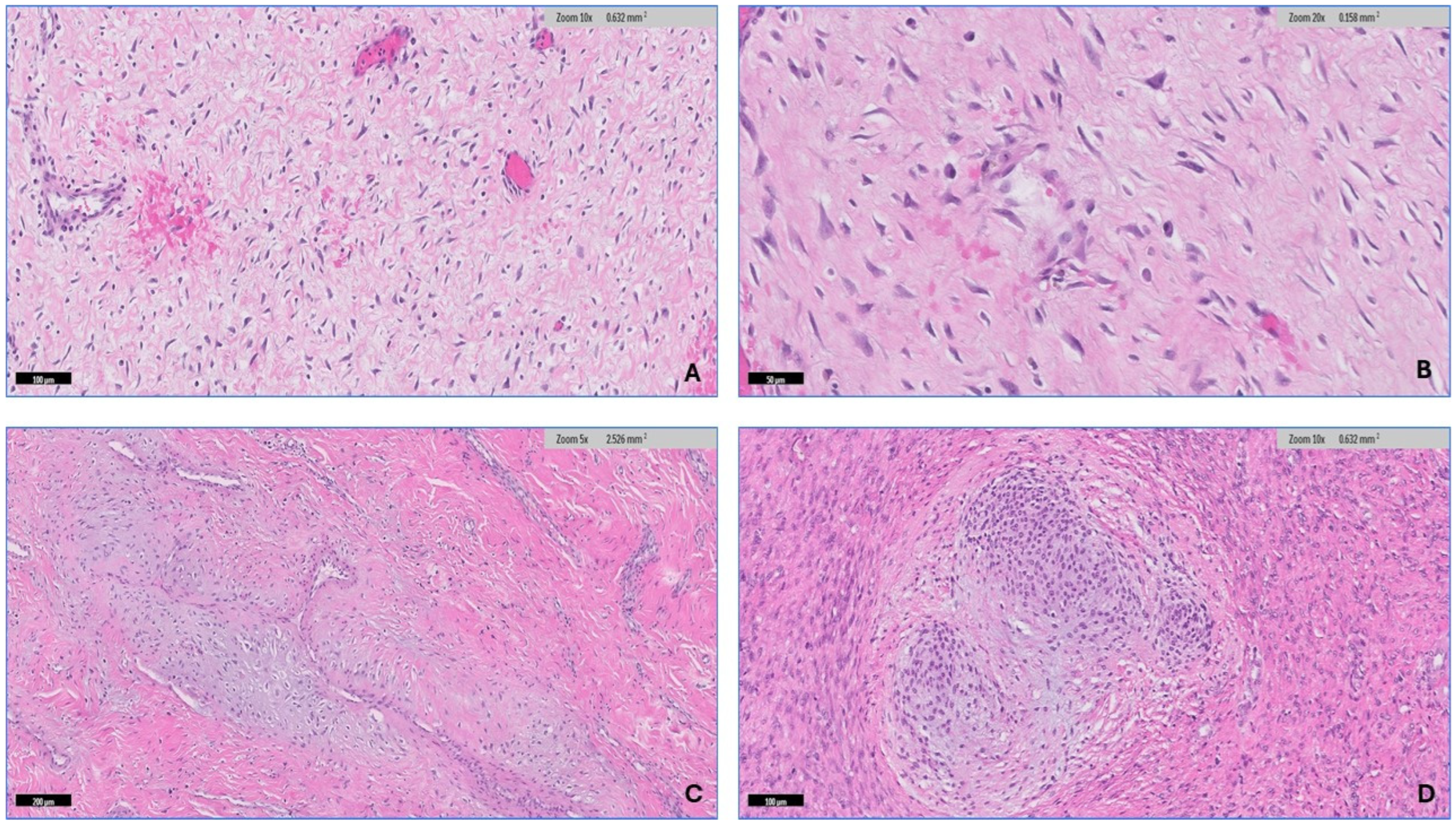
2.2. Microscopic Features
2.3. Immunohistochemistry
2.4. Next-Generation Sequencing (NGS) DNA and RNA Sequencing
3. Results
3.1. Clinical Features
3.2. Histopathologic Findings
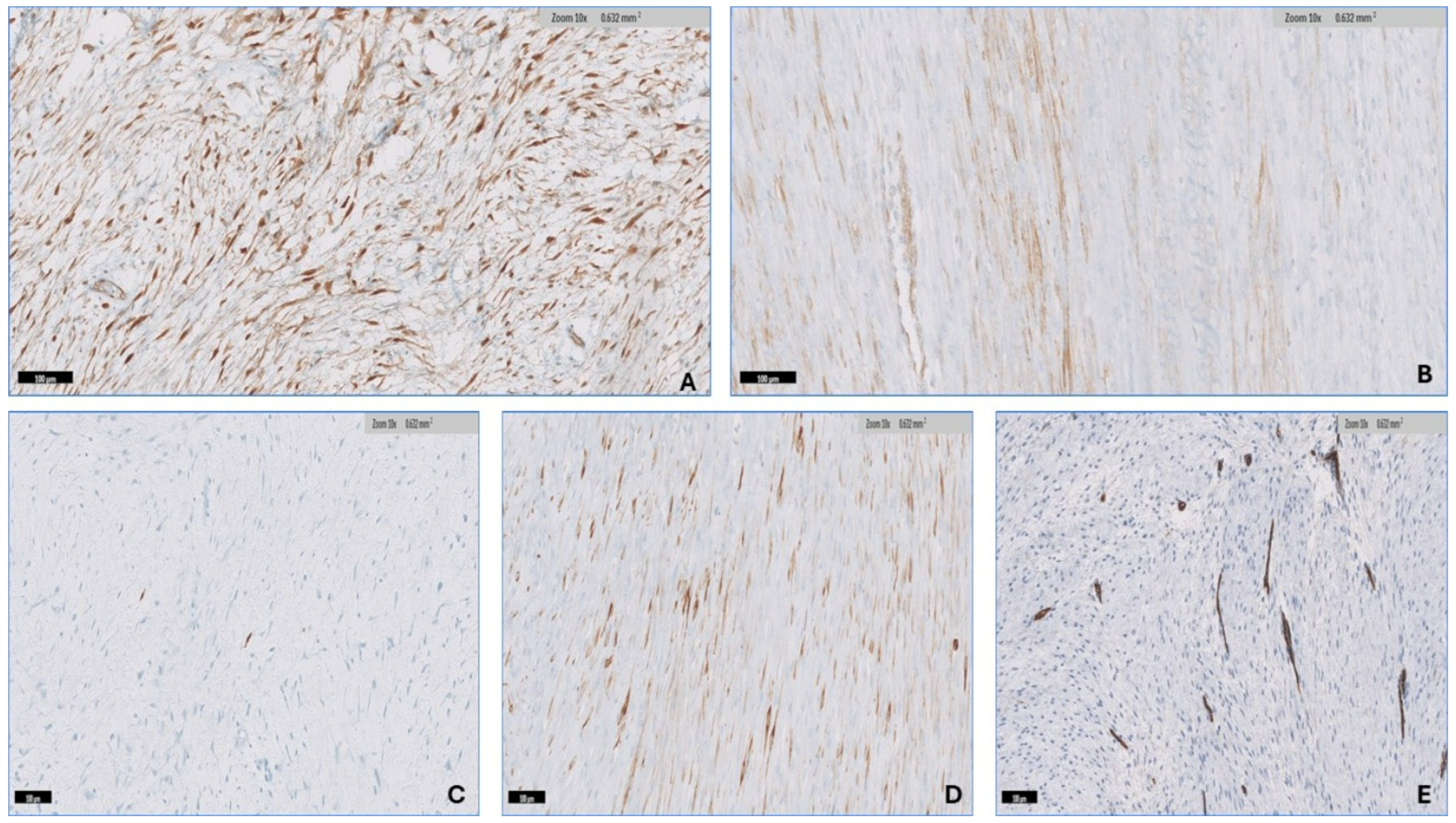
3.3. Immunohistochemistry
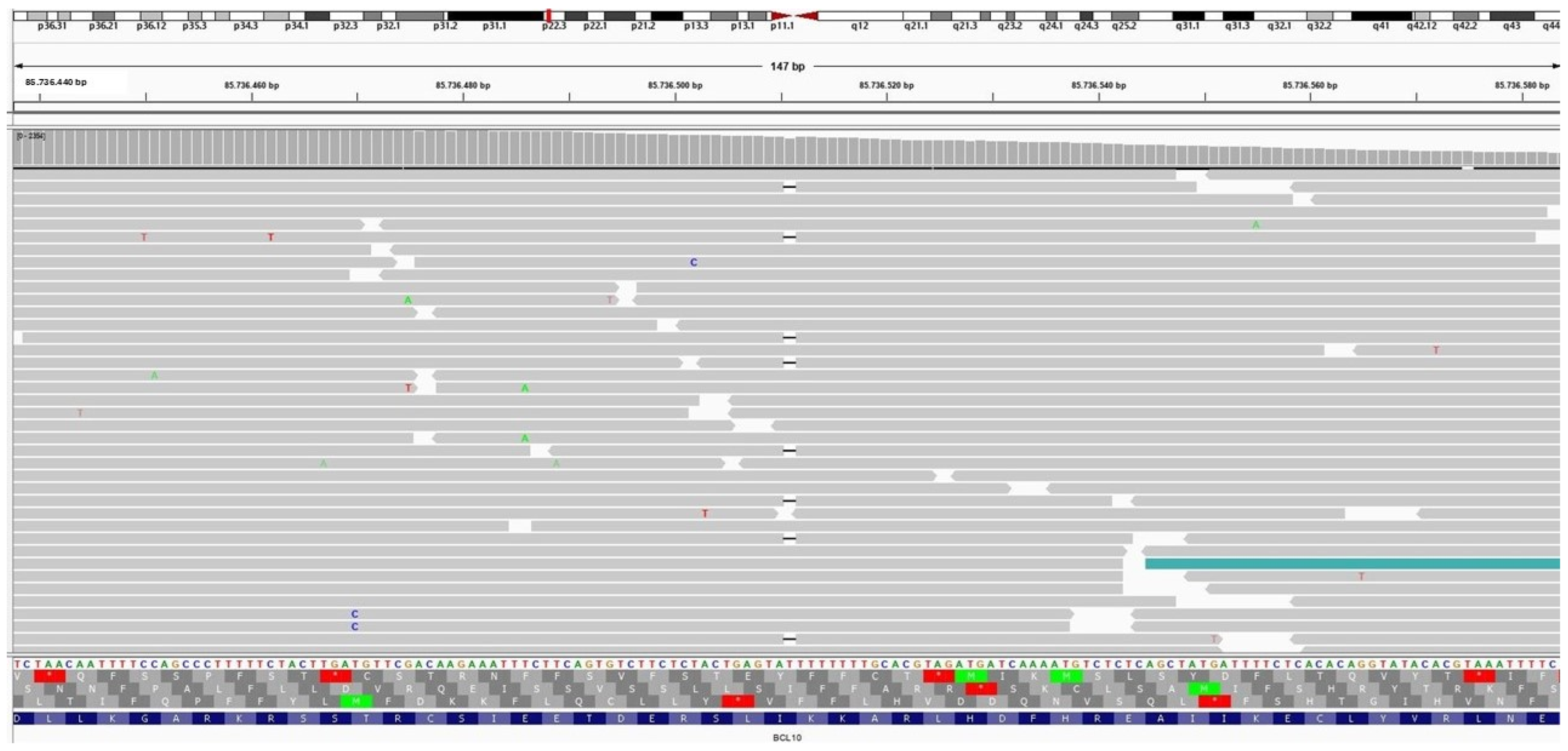
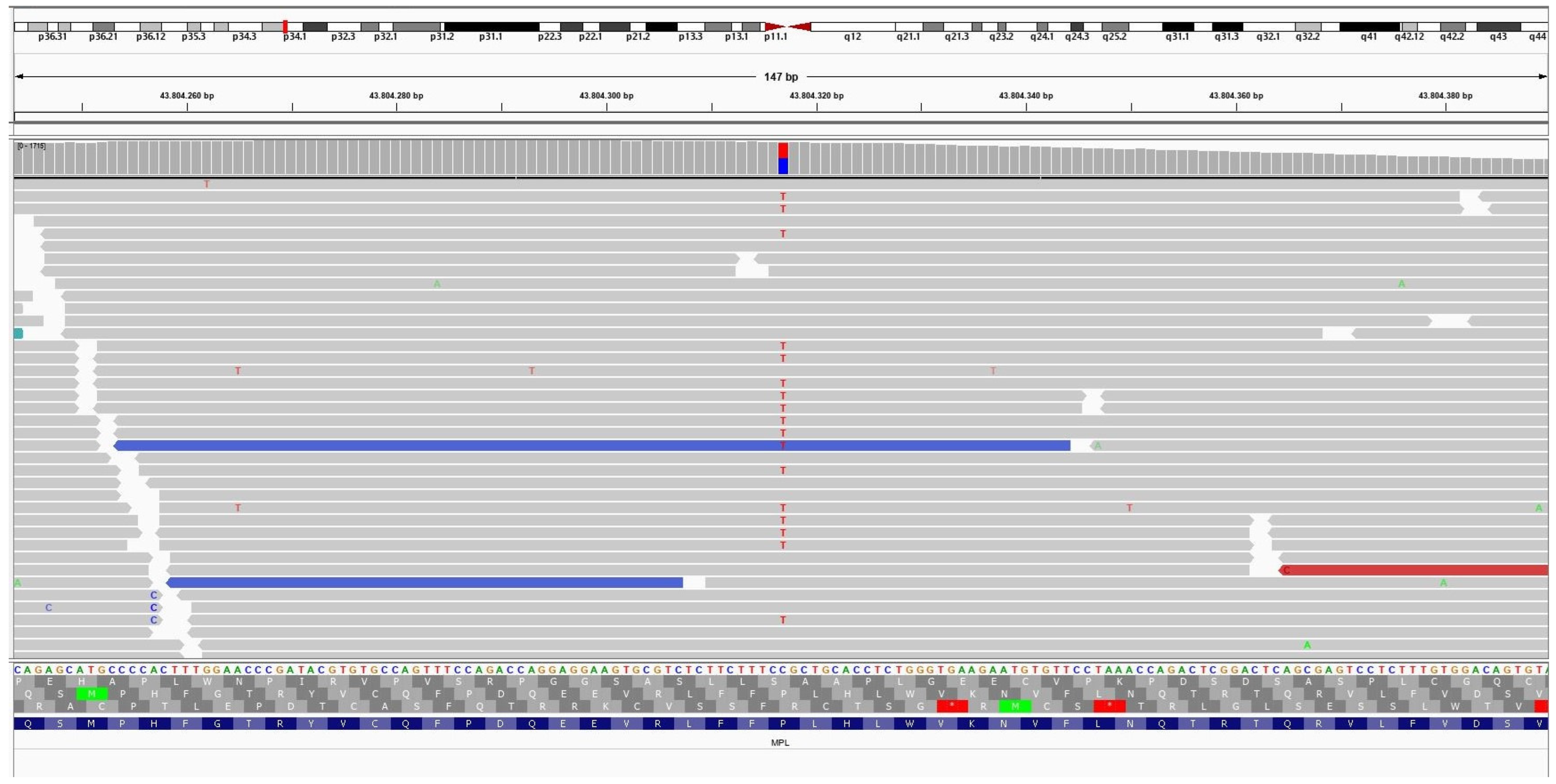
3.4. Molecular Findings
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 3. [Google Scholar]
- The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur. J. Cancer 2020, 127, 96–107. [CrossRef] [PubMed]
- Nieuwenhuis, M.H.; Casparie, M.; Mathus-Vliegen, L.M.; Dekkers, O.M.; Hogendoorn, P.C.; Vasen, H.F. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int. J. Cancer 2011, 129, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Baumgarten, C.; Garcia, J.; Bonvalot, S.; Haas, R.; Haller, F.; Hohenberger, P.; Penel, N.; Messiou, C.; van der Graaf, W.T.; et al. An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann. Oncol. 2017, 28, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Ströbel, P.; Hohenberger, P. Desmoid tumors: Clinical features and treatment options for advanced disease. Oncologist 2011, 16, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Penel, N.; Coindre, J.M.; Bonvalot, S.; Italiano, A.; Neuville, A.; Le Cesne, A.; Terrier, P.; Ray-Coquard, I.; Ranchere-Vince, D.; Robin, Y.M.; et al. Management of desmoid tumours: A nationwide survey of labelled reference centre networks in France. Eur. J. Cancer 2016, 58, 90–96. [Google Scholar] [CrossRef]
- van Broekhoven, D.L.; Grünhagen, D.J.; den Bakker, M.A.; van Dalen, T.; Verhoef, C. Time trends in the incidence and treatment of extra-abdominal and abdominal aggressive fibromatosis: A population-based study. Ann. Surg. Oncol. 2015, 22, 2817–2823. [Google Scholar] [CrossRef]
- Fiore, M.; MacNeill, A.; Gronchi, A.; Colombo, C. Desmoid-Type Fibromatosis: Evolving Treatment Standards. Surg. Oncol. Clin. N. Am. 2016, 25, 803–826. [Google Scholar] [CrossRef]
- de Camargo, V.P.; Keohan, M.L.; D’Adamo, D.R.; Antonescu, C.R.; Brennan, M.F.; Singer, S.; Ahn, L.S.; Maki, R.G. Clinical outcomes of systemic therapy for patients with deep fibromatosis (desmoid tumor). Cancer 2010, 116, 2258–2265. [Google Scholar] [CrossRef]
- Allen, P.W. The fibromatoses: A clinicopathologic classification based on 140 cases. Am. J. Surg. Pathol. 1977, 1, 255–270. [Google Scholar] [CrossRef]
- Fallen, T.; Wilson, M.; Morlan, B.; Lindor, N.M. Desmoid tumors—A characterization of patients seen at Mayo Clinic 1976–1999. Fam Cancer 2006, 5, 191–194. [Google Scholar] [CrossRef]
- Mehrotra, A.K.; Sheikh, S.; Aaron, A.D.; Montgomery, E.; Goldblum, J.R. Fibromatoses of the extremities: Clinicopathologic study of 36 cases. J. Surg. Oncol. 2000, 74, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Reitamo, J.J.; Häyry, P.; Nykyri, E.; Saxén, E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Am. J. Clin. Pathol. 1982, 77, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Sobin, L.H.; Shekitka, K.M.; Federspiel, B.H.; Helwig, E.B. Intra-abdominal fibromatosis. A pathologic analysis of 130 tumors with comparison of clinical subgroups. Am. J. Surg. Pathol. 1990, 14, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Zreik, R.T.; Fritchie, K.J. Morphologic Spectrum of Desmoid-Type Fibromatosis. Am. J. Clin. Pathol. 2016, 145, 332–340. [Google Scholar] [CrossRef]
- Timbergen, M.J.M.; Smits, R.; Grünhagen, D.J.; Verhoef, C.; Sleijfer, S.; Wiemer, E.A.C. Activated Signaling Pathways and Targeted Therapies in Desmoid-Type Fibromatosis: A Literature Review. Front. Oncol. 2019, 9, 397. [Google Scholar] [CrossRef]
- McLean, T.D.; Duchi, S.; Di Bella, C. Molecular Pathogenesis of Sporadic Desmoid Tumours and Its Implications for Novel Therapies: A Systematised Narrative Review. Target. Oncol. 2022, 17, 223–252. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, Q.; Liu, J.; Zhang, W. CTNNB1 Alternation Is a Potential Biomarker for Immunotherapy Prognosis in Patients With Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 759565. [Google Scholar] [CrossRef]
- AmeliMojarad, M.; AmeliMojarad, M.; Cui, X.; Shariati, P. Pan-cancer analysis of CTNNB1 with potential as a therapeutic target for human tumorigenesis. Inform. Med. Unlocked 2023, 42, 101331. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- Norkowski, E.; Masliah-Planchon, J.; Le Guellec, S.; Trassard, M.; Courrèges, J.B.; Charron-Barra, C.; Terrier, P.; Bonvalot, S.; Coindre, J.M.; Laé, M. Lower Rate of CTNNB1 Mutations and Higher Rate of APC Mutations in Desmoid Fibromatosis of the Breast: A Series of 134 Tumors. Am. J. Surg. Pathol. 2020, 44, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Enzo, M.V.; Rastrelli, M.; Rossi, C.R.; Hladnik, U.; Segat, D. The Wnt/β-catenin pathway in human fibrotic-like diseases and its eligibility as a therapeutic target. Mol. Cell Ther. 2015, 3, 1. [Google Scholar] [CrossRef]
- Ng, T.L.; Gown, A.M.; Barry, T.S.; Cheang, M.C.U.; Chan, A.K.W.; Turbin, D.A.; Hsu, F.D.; West, R.B.; Nielsen, T.O. Nuclear beta-catenin in mesenchymal tumors. Mod. Pathol. 2005, 18, 68–74. [Google Scholar] [CrossRef]
- Gurbuz, A.K.; Giardiello, F.M.; Petersen, G.M.; Krush, A.J.; Offerhaus, G.J.; Booker, S.V.; Kerr, M.C.; Hamilton, S.R. Desmoid tumours in familial adenomatous polyposis. Gut 1994, 35, 377–381. [Google Scholar] [CrossRef]
- Salas, S.; Chibon, F.; Noguchi, T.; Terrier, P.; Ranchere-Vince, D.; Lagarde, P.; Benard, J.; Forget, S.; Blanchard, C.; Dômont, J.; et al. Molecular characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes. Chromosomes Cancer 2010, 49, 560–568. [Google Scholar] [CrossRef]
- Koskenvuo, L.; Ristimäki, A.; Lepistö, A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J. Surg. Oncol. 2017, 116, 716–721. [Google Scholar] [CrossRef]
- Crago, A.M.; Chmielecki, J.; Rosenberg, M.; O’Connor, R.; Byrne, C.; Wilder, F.G.; Thorn, K.; Agius, P.; Kuk, D.; Socci, N.D.; et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer 2015, 54, 606–615. [Google Scholar] [CrossRef]
- Trautmann, M.; Rehkämper, J.; Gevensleben, H.; Becker, J.; Wardelmann, E.; Hartmann, W.; Grünewald, I.; Huss, S. Novel pathogenic alterations in pediatric and adult desmoid-type fibromatosis—A systematic analysis of 204 cases. Sci. Rep. 2020, 10, 3368. [Google Scholar] [CrossRef]
- Riedel, R.F.; Agulnik, M. Evolving strategies for management of desmoid tumor. Cancer 2022, 128, 3027–3040. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.; Thway, K. Aggressive fibromatosis. Pathology 2014, 46, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Reitamo, J.J.; Scheinin, T.M.; Häyry, P. The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the desmoid tumor. Am. J. Surg. 1986, 151, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Häyry, P.; Reitamo, J.J.; Tötterman, S.; Hopfner-Hallikainen, D.; Sivula, A. The Desmoid Tumor. II.: Analysis of Factors Possibly Contributing to the Etiology and Growth Behavior. Am. J. Clin. Pathol. 1982, 77, 674–680. [Google Scholar] [CrossRef]
- An, J.; Woo, H.Y.; Lee, Y.; Kim, H.S.; Jeong, J.; Kim, S.K. Clinicopathological features of 70 desmoid-type fibromatoses confirmed by β-catenin immunohistochemical staining and CTNNB1 mutation analysis. PLoS ONE 2021, 16, e0250619. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.U.; Din, N.U.; Abdul-Ghafar, J.; Park, Y.K. The many faces of solitary fibrous tumor; diversity of histological features, differential diagnosis and role of molecular studies and surrogate markers in avoiding misdiagnosis and predicting the behavior. Diagn. Pathol. 2021, 16, 32. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Rubin, B.P.; Kilpatrick, S.E.; Gjorgova Gjeorgjievski, S.; Fritchie, K.J.; Goldblum, J.R.; McKenney, J.K.; Billings, S.D. CD34-negative Solitary Fibrous Tumor: A Clinicopathologic Study of 25 Cases and Comparison with Their CD34-positive Counterparts. Am. J. Surg. Pathol. 2021, 45, 1616–1625. [Google Scholar] [CrossRef]
- Doyle, L.A.; Vivero, M.; Fletcher, C.D.; Mertens, F.; Hornick, J.L. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod. Pathol. 2014, 27, 390–395. [Google Scholar] [CrossRef]
- Kuba, M.G.; Lester, S.C.; Giess, C.S.; Bertagnolli, M.M.; Wieczorek, T.J.; Brock, J.E. Fibromatosis of the Breast: Diagnostic Accuracy of Core Needle Biopsy. Am. J. Clin. Pathol. 2017, 148, 243–250. [Google Scholar] [CrossRef]
- Lorenzen, J.; Cramer, M.; Buck, N.; Friedrichs, K.; Graubner, K.; Lühr, C.S.; Lindner, C.; Niendorf, A. Desmoid Type Fibromatosis of the Breast: Ten-Year Institutional Results of Imaging, Histopathology, and Surgery. Breast Care 2020, 16, 77–84. [Google Scholar] [CrossRef]
- Yoshida, A.; Tsuta, K.; Ohno, M.; Yoshida, M.; Narita, Y.; Kawai, A.; Asamura, H.; Kushima, R. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am. J. Surg. Pathol. 2014, 38, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Crago, A.M.; Rosenberg, M.; O’Connor, R.; Walker, S.R.; Ambrogio, L.; Auclair, D.; McKenna, A.; Heinrich, M.C.; Frank, D.A.; et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat. Genet. 2013, 45, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.-S.; Chen, C.-L.; Zhang, L.; Wang, R.; Su, F.; et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat. Genet. 2013, 45, 180–185. [Google Scholar] [CrossRef]
- Guseva, N.V.; Tanas, M.R.; Stence, A.A.; Sompallae, R.; Schade, J.C.; Bossler, A.D.; Bellizzi, A.M.; Ma, D. The NAB2–STAT6 gene fusion in solitary fibrous tumor can be reliably detected by anchored multiplexed PCR for targeted next-generation sequencing. Cancer Genet. 2016, 209, 303–312. [Google Scholar] [CrossRef]
- Demicco, E.G.; Harms, P.W.; Patel, R.M.; Smith, S.C.; Ingram, D.; Torres, K.; Carskadon, S.L.; Camelo-Piragua, S.; McHugh, J.B.; Siddiqui, J.; et al. Extensive survey of STAT6 expression in a large series of mesenchymal tumors. Am. J. Clin. Pathol. 2015, 143, 672–682. [Google Scholar] [CrossRef]
- Carlson, J.W.; Fletcher, C.D.M. Immunohistochemistry for β-catenin in the differential diagnosis of spindle cell lesions: Analysis of a series and review of the literature. Histopathology 2007, 51, 509–514. [Google Scholar] [CrossRef]
- Fisher, C. Immunohistochemistry in diagnosis of soft tissue tumours. Histopathology 2011, 58, 1001–1012. [Google Scholar] [CrossRef]
- Zhong, L.l.; Huang, G.x.; Xian, L.y.; Wei, Z.c.; Tang, Z.p.; Chen, Q.y.; Chen, H.; Tang, F. Novel characteristics for immunophenotype, FISH pattern and molecular cytogenetics in synovial sarcoma. Sci. Rep. 2023, 13, 7954. [Google Scholar] [CrossRef] [PubMed]
- Lasota, J.; Chłopek, M.; Kaczorowski, M.; Natálie, K.; Ryś, J.; Kopczyński, J.; Sulaieva, O.; Michal, M.; Kruczak, A.; Harazin-Lechowska, A.; et al. Utility of Immunohistochemistry With Antibodies to SS18-SSX Chimeric Proteins and C-Terminus of SSX Protein for Synovial Sarcoma Differential Diagnosis. Am. J. Surg. Pathol. 2024, 48, 97–105. [Google Scholar] [CrossRef]
- Nishio, J.; Nakayama, S.; Nabeshima, K.; Yamamoto, T. Biology and Management of Dedifferentiated Liposarcoma: State of the Art and Perspectives. J. Clin. Med. 2021, 10, 3230. [Google Scholar] [CrossRef]
- Kimura, H.; Dobashi, Y.; Nojima, T.; Nakamura, H.; Yamamoto, N.; Tsuchiya, H.; Ikeda, H.; Sawada-Kitamura, S.; Oyama, T.; Ooi, A. Utility of fluorescence in situ hybridization to detect MDM2 amplification in liposarcomas and their morphological mimics. Int. J. Clin. Exp. Pathol. 2013, 6, 1306–1316. [Google Scholar] [PubMed]
- Belakhoua, S.M.; Rodriguez, F.J. Diagnostic Pathology of Tumors of Peripheral Nerve. Neurosurgery 2021, 88, 443–456. [Google Scholar] [CrossRef]
- Robson, A.; Allen, P.; Hollowood, K. S100 expression in cutaneous scars: A potential diagnostic pitfall in the diagnosis of desmoplastic melanoma. Histopathology 2001, 38, 135–140. [Google Scholar] [CrossRef]
- Brčić, I.; Godschachner, T.M.; Bergovec, M.; Igrec, J.; Till, H.; Lackner, H.; Scheipl, S.; Kashofer, K.; Brodowicz, T.; Leithner, A.; et al. Broadening the spectrum of NTRK rearranged mesenchymal tumors and usefulness of pan-TRK immunohistochemistry for identification of NTRK fusions. Mod. Pathol. 2021, 34, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Siozopoulou, V.; Smits, E.; De Winne, K.; Marcq, E.; Pauwels, P. NTRK Fusions in Sarcomas: Diagnostic Challenges and Clinical Aspects. Diagnostics 2021, 11, 478. [Google Scholar] [CrossRef]
- Lao, I.W.; Sun, M.; Zhao, M.; Yu, L.; Wang, J. Lipofibromatosis-like neural tumour: A clinicopathological study of ten additional cases of an emerging novel entity. Pathology 2018, 50, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; Gibson, S.; Ramsay, A.; Sebire, N.J. Beta-catenin expression in pediatric fibroblastic and myofibroblastic lesions: A study of 100 cases. Pediatr. Dev. Pathol. 2009, 12, 292–296. [Google Scholar] [CrossRef]
- Dachy, G.; de Krijger, R.R.; Fraitag, S.; Théate, I.; Brichard, B.; Hoffman, S.B.; Libbrecht, L.; Arts, F.A.; Brouillard, P.; Vikkula, M.; et al. Association of PDGFRB Mutations With Pediatric Myofibroma and Myofibromatosis. JAMA Dermatol. 2019, 155, 946–950. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Sung, Y.S.; Zhang, L.; Agaram, N.P.; Fletcher, C.D. Recurrent SRF-RELA Fusions Define a Novel Subset of Cellular Myofibroma/Myopericytoma: A Potential Diagnostic Pitfall With Sarcomas With Myogenic Differentiation. Am. J. Surg. Pathol. 2017, 41, 677–684. [Google Scholar] [CrossRef]
- Owens, C.L.; Sharma, R.; Ali, S.Z. Deep fibromatosis (desmoid tumor). Cancer 2007, 111, 166–172. [Google Scholar] [CrossRef]
- Cessna, M.H.; Zhou, H.; Perkins, S.L.; Tripp, S.R.; Layfield, L.; Daines, C.; Coffin, C.M. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am. J. Surg. Pathol. 2001, 25, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Tos, A.P.D.; Hornick, J.L.; Miettinen, M.; WHO Classification of Tumours Editorial Board. Digestive System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019; Volume 1. [Google Scholar]
- Hornick, J.L. Novel uses of immunohistochemistry in the diagnosis and classification of soft tissue tumors. Mod. Pathol. 2014, 27, S47–S63. [Google Scholar] [CrossRef] [PubMed]
- Dow, N.; Giblen, G.; Sobin, L.H.; Miettinen, M. Gastrointestinal stromal tumors: Differential diagnosis. Semin. Diagn. Pathol. 2006, 23, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Odintsov, I.; Dong, F.; Guenette, J.P.; Fritchie, K.J.; Jo, V.Y.; Fletcher, C.D.M.; Papke, D.J., Jr. Infantile Sinonasal Myxoma Is Clinically and Genetically Distinct From Other Myxomas of the Craniofacial Bones and From Desmoid Fibromatosis. Am. J. Surg. Pathol. 2023, 47, 1301–1315. [Google Scholar] [CrossRef]
- Kuhnen, C.; Herter, P.; Müller, O.; Muehlberger, T.; Krause, L.; Homann, H.; Steinau, H.U.; Müller, K.-M. β-Catenin in Soft tissue Sarcomas: Expression is Related to Proliferative Activity in High-Grade Sarcomas. Mod. Pathol. 2000, 13, 1005–1013. [Google Scholar] [CrossRef]
- Ronen, S.; Ko, J.S.; Rubin, B.P.; Kilpatrick, S.E.; Wang, W.L.; Lazar, A.J.; Goldblum, J.R.; Billings, S.D. Superficial low-grade fibromyxoid sarcoma. J. Cutan. Pathol. 2023, 50, 147–154. [Google Scholar] [CrossRef]
- Reid, R.; de Silva, M.V.; Paterson, L.; Ryan, E.; Fisher, C. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes share a common t(7;16)(q34;p11) translocation. Am. J. Surg. Pathol. 2003, 27, 1229–1236. [Google Scholar] [CrossRef]
- Doyle, L.A.; Möller, E.; Dal Cin, P.; Fletcher, C.D.M.; Mertens, F.; Hornick, J.L. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am. J. Surg. Pathol. 2011, 35, 733–741. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Dilworth, H.P.; Iacobuzio-Donahue, C.; Ricci, F.; Weber, K.; Furlong, M.A.; Fisher, C.; Montgomery, E. Nuclear beta-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am. J. Surg. Pathol. 2005, 29, 653–659. [Google Scholar] [CrossRef]
- Erickson-Johnson, M.R.; Chou, M.M.; Evers, B.R.; Roth, C.W.; Seys, A.R.; Jin, L.; Ye, Y.; Lau, A.W.; Wang, X.; Oliveira, A.M. Nodular fasciitis: A novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab. Investig. 2011, 91, 1427–1433. [Google Scholar] [CrossRef]
- Meazza, C.; Belfiore, A.; Busico, A.; Settanni, G.; Paielli, N.; Cesana, L.; Ferrari, A.; Chiaravalli, S.; Massimino, M.; Gronchi, A.; et al. AKT1 and BRAF mutations in pediatric aggressive fibromatosis. Cancer Med. 2016, 5, 1204–1213. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Clone | Source | Specific Antibody Concentration |
|---|---|---|---|
| β-catenin | β-catenin (14) | Ventana, REF 760-4242 | 2 µg/mL |
| CD34 | CONFIRM Anti-CD34 (QBEnd/10) | Ventana, REF 790-2927 | 0.8 µg/mL |
| Desmin | CONFIRM Anti-desmin (DE-R-11) | Ventana, REF 760-2513 | 5 µg/mL |
| SMA | Actin, smooth muscle (1A4) | Ventana, 760-2833 | 0.03 µg/mL |
| S100 | CONFIRM anti-S100 (4C4.9) | Ventana, REF 790-2914 | 10 µg/mL |
| DTF Anatomical Classification N = 33 | DTF Specific Site | Number of Specimen |
|---|---|---|
| Abdominal wall | Abdominal wall | 6 specimens, 18.18% |
| Intra-abdominal | 8 specimens, 24.24% | |
| Mesentery | 5 | |
| Pelvic mass | 3 | |
| Extra-abdominal | 19 specimens, 57.58% | |
| Left mandible | 1 | |
| Right lateral | 1 | |
| pharyngeal wall | 1 | |
| Right breast | 1 | |
| Left breast | 1 | |
| Left thigh | 1 | |
| Right shoulder | 1 | |
| Left side back | 1 | |
| Right neck | 1 | |
| Right shoulder mass subscapular | 1 | |
| Left shoulder | 1 | |
| Right groin | 1 | |
| Right parotid | 1 | |
| Central back of neck | 1 | |
| Back | 1 | |
| Submental | 1 | |
| Left axilla | 1 | |
| Right thigh | 1 | |
| Right leg | 1 |
| The Mean of Histologic Pattern | Patients with Recurrence N = 7 | Patients with no Recurrence N = 11 | p Value |
|---|---|---|---|
| Conventional pattern | 46% | 63% | 0.299 |
| Hypercellular pattern | 9% | 12% | 0.5413 |
| Myxoid pattern | 16.42% | 10% | 0.3673 |
| Hyalinized/Hypocellular patten | 24.28% | 6.36% | 0.0895 |
| Staghorn/hemangiopericytomatous blood vessels pattern | 4.28% | 7.27% | 0.3922 |
| Nodular fasciitis-like pattern | 2.85% | 6.81% | 0.5738 |
| Keloid-like pattern | 2.14% | 2.72% | 0.8247 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajabnoor, R. Different Shades of Desmoid-Type Fibromatosis (DTF): Detection of Noval Mutations in the Clinicopathologic Analysis of 32 Cases. Diagnostics 2024, 14, 2161. https://doi.org/10.3390/diagnostics14192161
Ajabnoor R. Different Shades of Desmoid-Type Fibromatosis (DTF): Detection of Noval Mutations in the Clinicopathologic Analysis of 32 Cases. Diagnostics. 2024; 14(19):2161. https://doi.org/10.3390/diagnostics14192161
Chicago/Turabian StyleAjabnoor, Rana. 2024. "Different Shades of Desmoid-Type Fibromatosis (DTF): Detection of Noval Mutations in the Clinicopathologic Analysis of 32 Cases" Diagnostics 14, no. 19: 2161. https://doi.org/10.3390/diagnostics14192161
APA StyleAjabnoor, R. (2024). Different Shades of Desmoid-Type Fibromatosis (DTF): Detection of Noval Mutations in the Clinicopathologic Analysis of 32 Cases. Diagnostics, 14(19), 2161. https://doi.org/10.3390/diagnostics14192161