

Enhancing Influenza Detection through Integrative Machine Learning and Nasopharyngeal Metabolomic Profiling: A Comprehensive Study

 , , and
, , and 
Abstract
1. Introduction
- This study utilized a nasopharyngeal metabolomics dataset that was publicly available, as reported by Hogan et al. [28], ensuring transparency and accessibility in data sourcing.
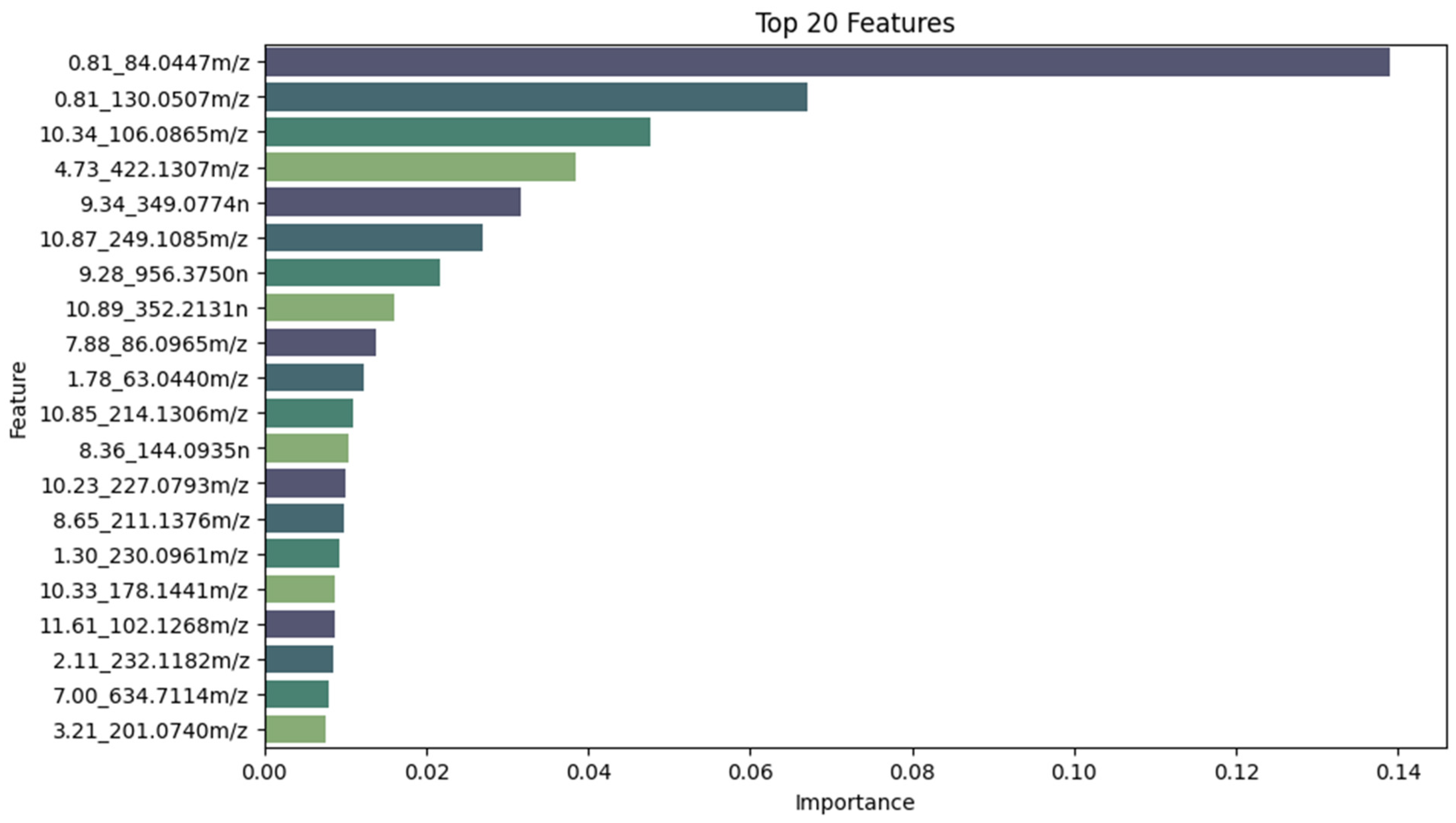
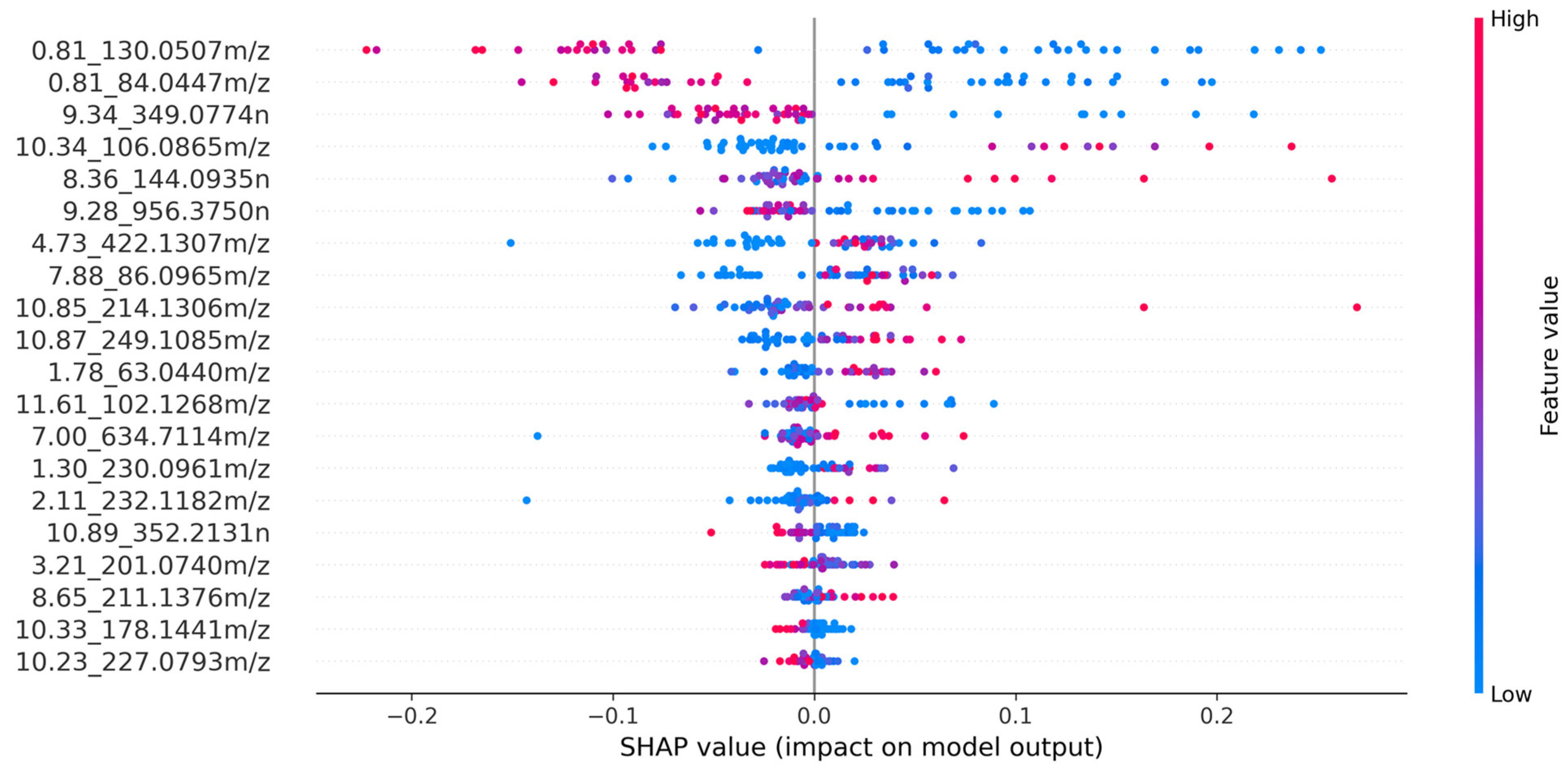
- The top 20 features were selected using Shapley Additive Explanations (SHAP) values.
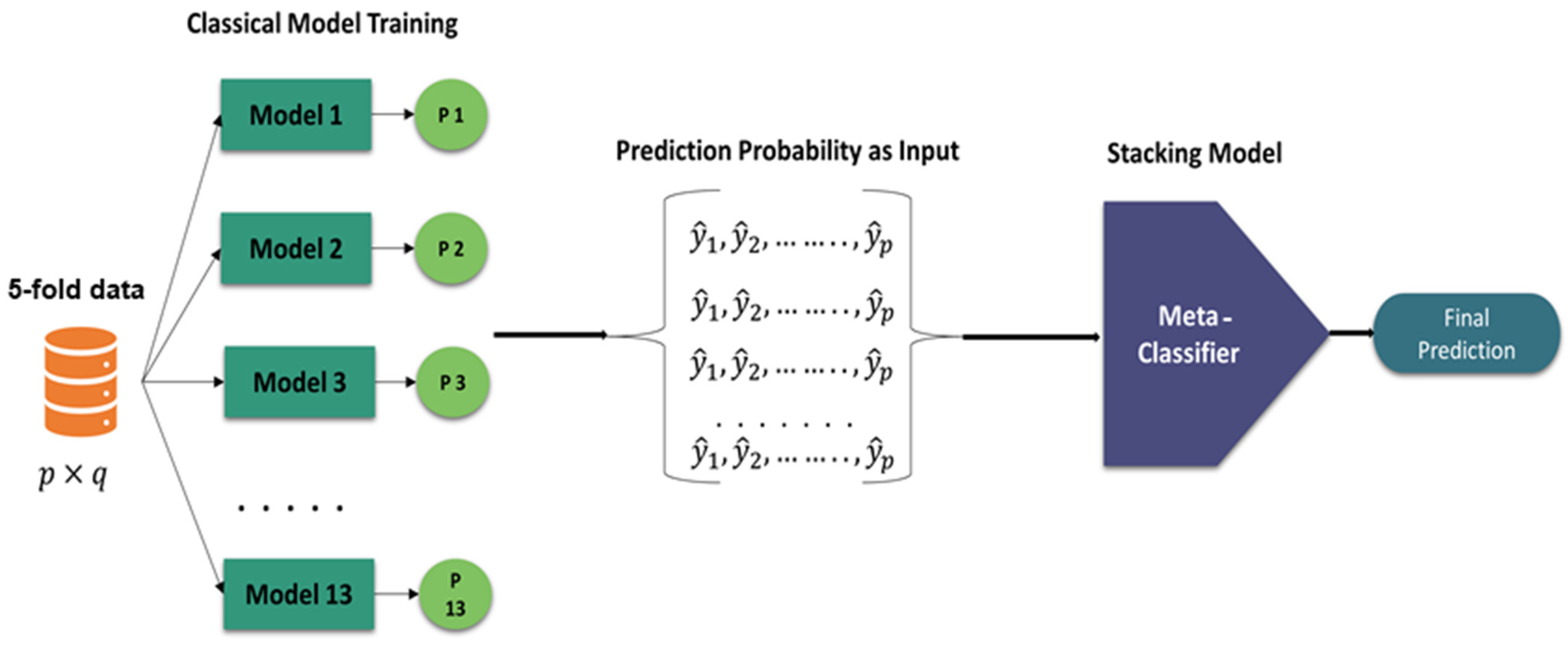
- A stacking-based meta-classifier approach was proposed with 5-fold cross-validation. Initially, thirteen machine learning models were trained, and the three best models were selected based on their probabilities. These three models were combined to train 13 meta-classifier models.
- For the primary dataset, the ExtraTrees stacking model achieved the best results with 97.08% accuracy. External validation yielded 100% accuracy.
- Model explainability was utilized to identify the biomarkers responsible for influenza prediction.
2. Methods and Materials
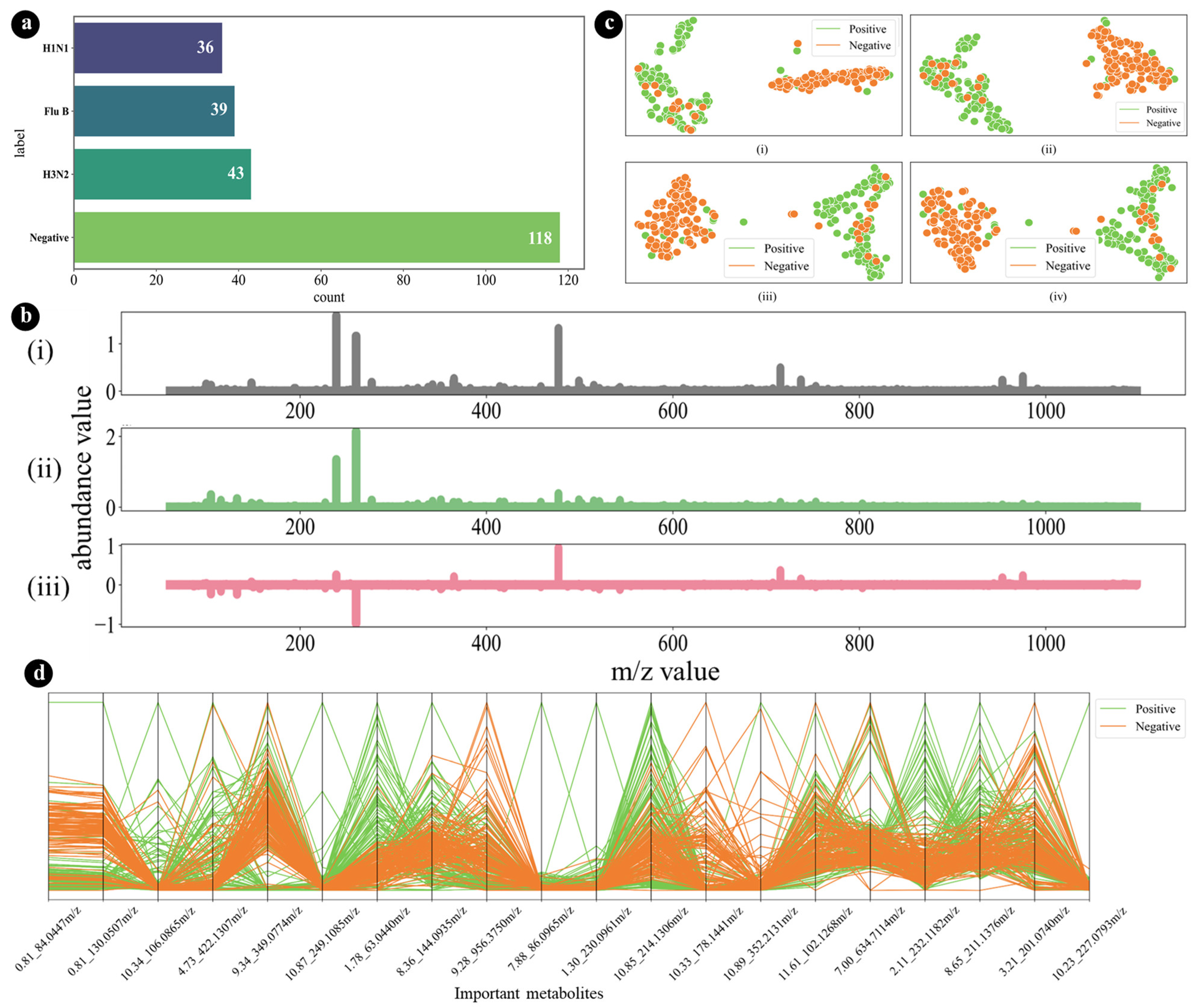
2.1. Description of the Dataset
2.2. Dataset Preprocessing
2.2.1. Dataset Cleaning
2.2.2. Missing Data Imputation
2.2.3. Dataset Splitting
2.2.4. Normalization
2.2.5. Feature Selection
2.3. Development of Machine Learning Models
Stacking-Based Meta-Classifier
2.4. Experimental Setup
2.5. Performance Metrics
3. Results
3.1. Internal Validation
3.2. External Validation
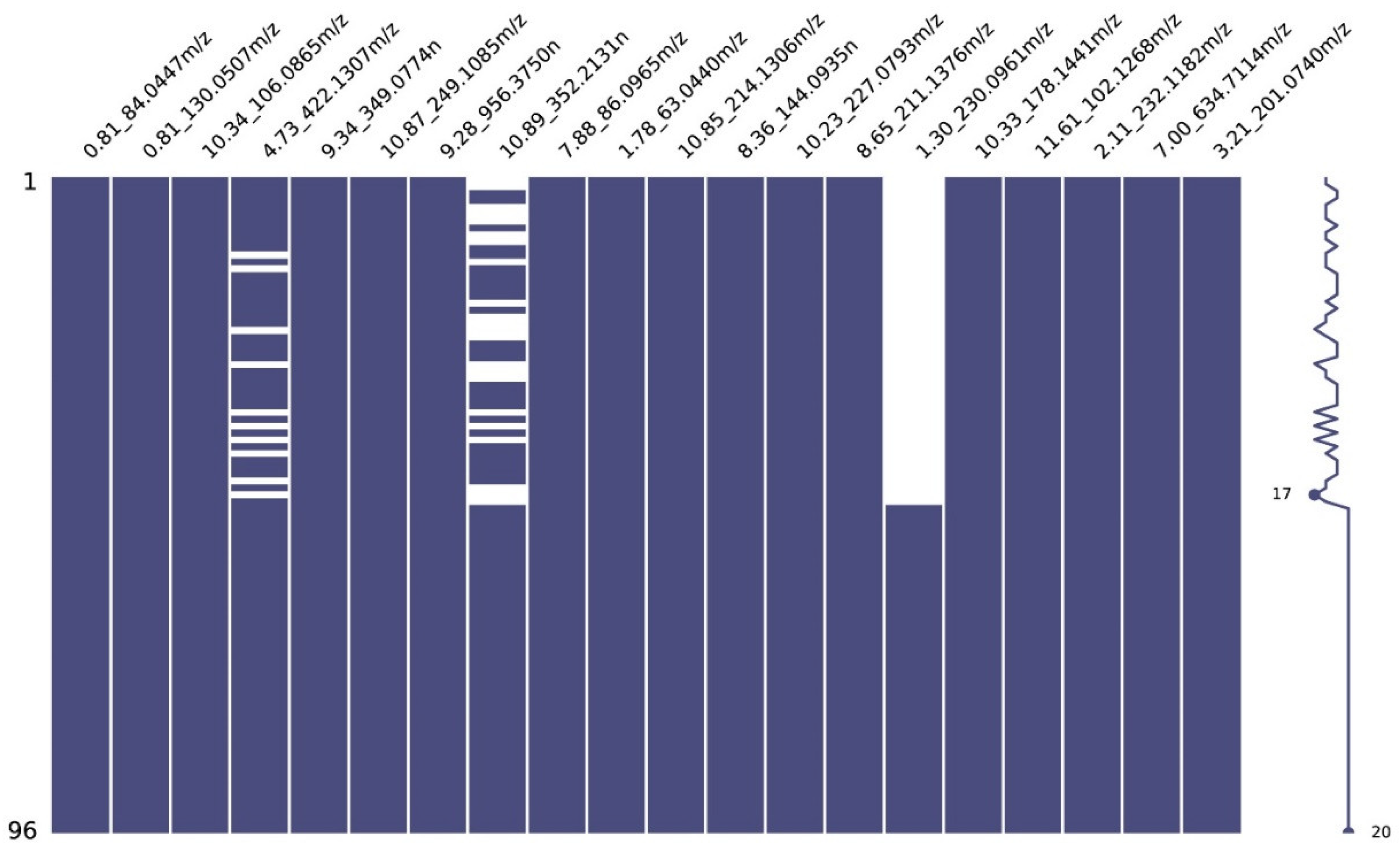
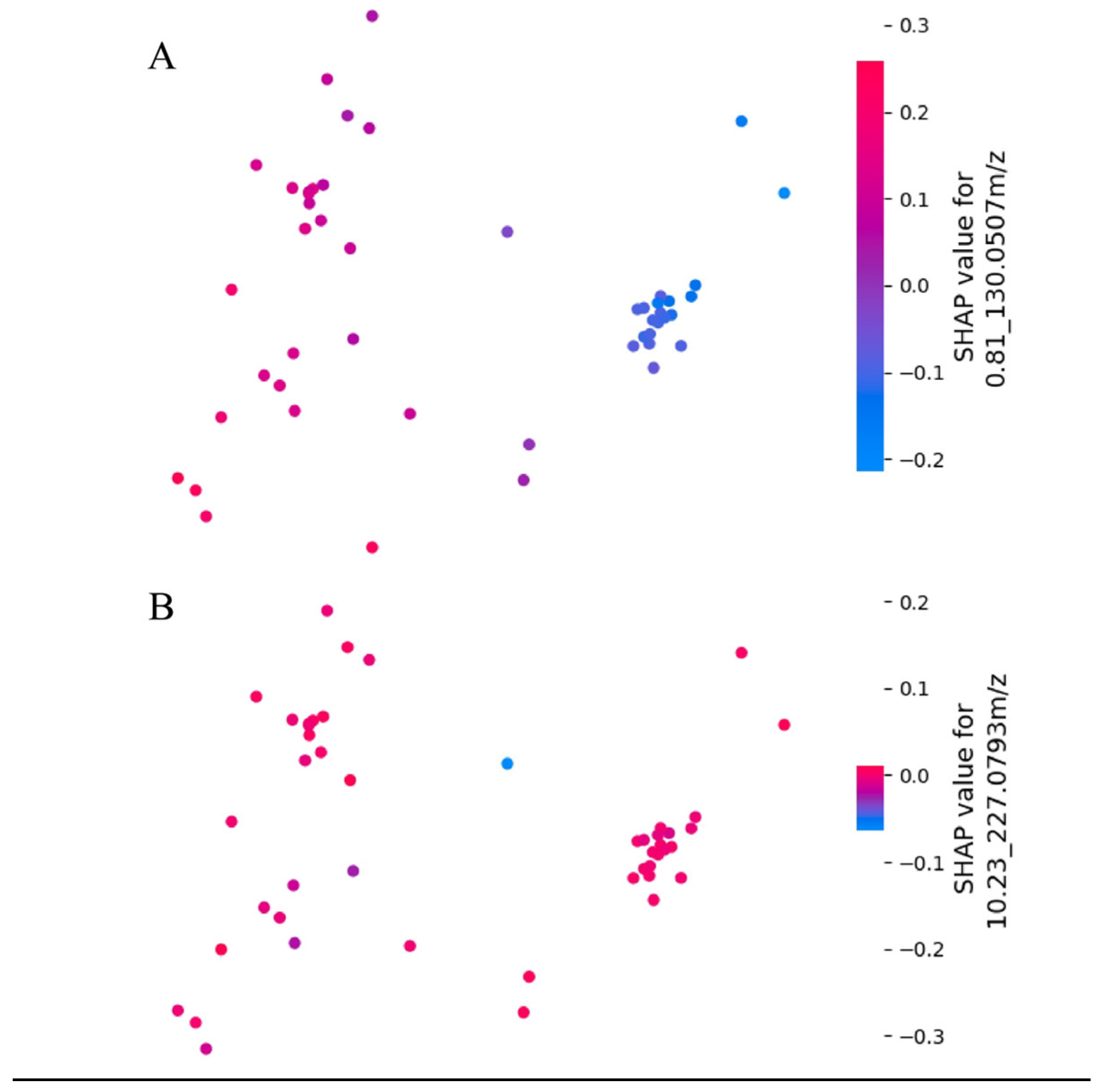
3.3. Model Explainability
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APACHE II | Acute Physiology and Chronic Health Evaluation II |
| AUC | area under the curve |
| COVID-19 | coronavirus disease 2019 |
| GC/MS | gas chromatography–mass spectrometry |
| HCC | hepatocellular carcinoma |
| IAVs | Influenza A viruses |
| IOKR | Input–Output Kernel Regression |
| LC/MS | liquid chromatography–mass spectrometry |
| LC/Q-TOF | liquid chromatography–quadrupole time-of-flight mass spectrometry |
| LGBM | light gradient-boosting machine |
| ML | machine learning |
| PCA | Principal Component Analysis |
| PCR | polymerase chain reaction |
| RF | Random Forest |
| RSV | respiratory syncytial virus |
| SHAP | Shapley Additive Explanations |
| SWQS | weighted quantile sketch |
| T-SNE | t-distributed Stochastic Neighbor Embedding |
| WHO | World Health Organization |
References
- Moghadami, M. A narrative review of influenza: A seasonal and pandemic disease. Iran. J. Med. Sci. 2017, 42, 2. [Google Scholar]
- WHO. Influenza. 2018. Available online: https://www.who.int/teams/health-product-policy-and-standards/standards-and-specifications/norms-and-standards/vaccine-standardization/influenza (accessed on 12 December 2023).
- Schmolke, M.; García-Sastre, A. Evasion of innate and adaptive immune responses by influenza A virus. Cell. Microbiol. 2010, 12, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Vossen, M.T.; Westerhout, E.M.; Söderberg-Nauclér, C.; Wiertz, E.J. Viral immune evasion: A masterpiece of evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Peteranderl, C.; Herold, S.; Schmoldt, C. Human influenza virus infections. In Proceedings of Seminars in Respiratory and Critical Care Medicine; Thieme Medical Publishers: New York, NY, USA, 2016; pp. 487–500. [Google Scholar]
- WHO. WHO Guidelines for Pharmacological Management of Pandemic Influenza A (H1N1) 2009 and Other Influenza Viruses: Part II Review of Evidence; WHO: Geneva, Switzerland, 2010; p. 61. [Google Scholar]
- Merckx, J.; Wali, R.; Schiller, I.; Caya, C.; Gore, G.C.; Chartrand, C.; Dendukuri, N.; Papenburg, J. Diagnostic accuracy of novel and traditional rapid tests for influenza infection compared with reverse transcriptase polymerase chain reaction: A systematic review and meta-analysis. Ann. Intern. Med. 2017, 167, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Somerville, L.K.; Ratnamohan, V.M.; Dwyer, D.E.; Kok, J. Molecular diagnosis of respiratory viruses. Pathology 2015, 47, 243–249. [Google Scholar] [CrossRef]
- Vergara, A.; Cilloniz, C.; Luque, N.; Garcia-Vidal, C.; Tejero, J.; Perelló, R.; Lucena, C.M.; Torres, A.; Marcos, M.A. Detection of human cytomegalovirus in bronchoalveolar lavage of intensive care unit patients. Eur. Respir. J. 2018, 51, 1701332. [Google Scholar] [CrossRef]
- Tan, S.K.; Burgener, E.B.; Waggoner, J.J.; Gajurel, K.; Gonzalez, S.; Chen, S.F.; Pinsky, B.A. Molecular and Culture-Based Bronchoalveolar Lavage Fluid Testing for the Diagnosis of Cytomegalovirus Pneumonitis. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Nicholson, J.K.; Wilson, I.D. Understanding Global Systems Biology: Metabonomics and the Continuum of Metabolism. Nat. Rev. Drug Discov. 2003, 2, 668–676. [Google Scholar] [CrossRef]
- Tounta, V.; Liu, Y.; Cheyne, A.; Larrouy-Maumus, G. Metabolomics in infectious diseases and drug discovery. Mol. Omics 2021, 17, 376–393. [Google Scholar] [CrossRef]
- Banoei, M.M.; Vogel, H.J.; Weljie, A.M.; Kumar, A.; Yende, S.; Angus, D.C.; Winston, B.W.; the Canadian Critical Care Translational Biology Group (CCCTBG). Plasma metabolomics for the diagnosis and prognosis of H1N1 influenza pneumonia. Crit. Care 2017, 21, 97. [Google Scholar] [CrossRef]
- Al-Sulaiti, H.; Almaliti, J.; Naman, C.B.; Al Thani, A.A.; Yassine, H.M. Metabolomics approaches for the diagnosis, treatment, and better disease management of viral infections. Metabolites 2023, 13, 948. [Google Scholar] [CrossRef]
- Beale, D.J.; Oh, D.Y.; Karpe, A.V.; Tai, C.; Dunn, M.S.; Tilmanis, D.; Palombo, E.A.; Hurt, A.C. Untargeted metabolomics analysis of the upper respiratory tract of ferrets following influenza A virus infection and oseltamivir treatment. Metabolomics 2019, 15, 33. [Google Scholar] [CrossRef] [PubMed]
- Humes, S.T.; Iovine, N.; Prins, C.; Garrett, T.J.; Lednicky, J.A.; Coker, E.S.; Sabo-Attwood, T. Association between lipid profiles and viral respiratory infections in human sputum samples. Respir. Res. 2022, 23, 177. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.A.; Klechka, L.; Hossain, E.; Parab, A.R.; Eaton, K.; Hinsdale, M.; McCall, L.-I. Spatial metabolomics reveals localized impact of influenza virus infection on the lung tissue metabolome. Msystems 2022, 7, e00353–e00422. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.B.; Chng, C.; Guan, X.L.; Lei, Z.; Rozen, S.G.; Wenk, M.R. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res. 2014, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Yassine, H.M.; Benslimane, F.M.; Smatti, M.K.; Schuchardt, S.; Albagha, O.; Al-Thani, A.A.; Ait Hssain, A.; Diboun, I.; Elrayess, M.A. Predictive biomarkers of intensive care unit and mechanical ventilation duration in critically-ill coronavirus disease 2019 patients. Front. Med. 2021, 8, 733657. [Google Scholar] [CrossRef] [PubMed]
- Wendt, C.H.; Castro-Pearson, S.; Proper, J.; Pett, S.; Griffin, T.J.; Kan, V.; Carbone, J.; Koulouris, N.; Reilly, C.; Neaton, J.D. Metabolite profiles associated with disease progression in influenza infection. PLoS ONE 2021, 16, e0247493. [Google Scholar] [CrossRef]
- Siddiqa, A.; Wang, Y.; Thapa, M.; Martin, D.E.; Cadar, A.N.; Bartley, J.M.; Li, S. A pilot metabolomic study of drug interaction with the immune response to seasonal influenza vaccination. NPJ Vaccines 2023, 8, 92. [Google Scholar] [CrossRef]
- Pacchiarotta, T.; Deelder, A.M.; Mayboroda, O.A. Metabolomic investigations of human infections. Bioanalysis 2012, 4, 919–925. [Google Scholar] [CrossRef]
- Zurfluh, S.; Baumgartner, T.; Meier, M.A.; Ottiger, M.; Voegeli, A.; Bernasconi, L.; Neyer, P.; Mueller, B.; Schuetz, P. The role of metabolomic markers for patients with infectious diseases: Implications for risk stratification and therapeutic modulation. Expert Rev. Anti-Infect. Ther. 2018, 16, 133–142. [Google Scholar] [CrossRef]
- Chi, N.F.; Chang, T.H.; Lee, C.Y.; Wu, Y.W.; Shen, T.A.; Chan, L.; Chen, Y.R.; Chiou, H.Y.; Hsu, C.Y.; Hu, C.J. Untargeted metabolomics predicts the functional outcome of ischemic stroke. J. Formos. Med. Assoc. 2021, 120, 234–241. [Google Scholar] [CrossRef]
- Delafiori, J.; Delafiori, J.; Navarro, L.C.; Siciliano, R.F.; de Melo, G.C.; Busanello, E.N.B.; Nicolau, J.C.; Sales, G.M.; de Oliveira, A.N.; Val, F.F.A.; et al. COVID-19 automated diagnosis and risk assessment through metabolomics and machine learning. Anal. Chem. 2021, 93, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Baiges-Gaya, G.; Iftimie, S.; Castae, H.; Rodriguez-Tomas, E.; Jimenez-Franco, A.; Lopez-Azcona, A.F.; Castro, A.; Camps, J.; Joven, J. Combining semi-targeted metabolomics and machine learning to identify metabolic alterations in the serum and urine of hospitalized patients with COVID-19. Biomolecules 2023, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Nitsch-Osuch, A.; Kuchar, E.; Golkebiak, I.; Kanecki, K.; Tarka, P.; Brydak, L.B. Rapid influenza diagnostic tests improve suitability of antiviral treatment in hospitalized children. Influenza Respir. Care 2017, 968, 1–6. [Google Scholar]
- Hogan, C.A.; Rajpurkar, P.; Sowrirajan, H.; Phillips, N.A.; Le Anthony, T.; Wu, M.; Garamani, N.; Sahoo, M.K.; Wood, M.L.; Huang, C.H.; et al. Nasopharyngeal metabolomics and machine learning approach for the diagnosis of influenza. eBioMedicine 2021, 71, 103546. [Google Scholar] [CrossRef]
- Mangalathu, S.; Hwang, S.-H.; Jeon, J.-S. Failure mode and effects analysis of RC members based on machine-learning-based SHapley Additive exPlanations (SHAP) approach. Eng. Struct. 2020, 219, 110927. [Google Scholar] [CrossRef]
- Ekanayake, I.; Meddage, D.; Rathnayake, U. A novel approach to explain the black-box nature of machine learning in compressive strength predictions of concrete using Shapley additive explanations (SHAP). Case Stud. Constr. Mater. 2022, 16, e01059. [Google Scholar] [CrossRef]
- Maćkiewicz, A.; Ratajczak, W. Principal Components Analysis (PCA). Comput. Geosci. 1993, 19, 303–342. [Google Scholar] [CrossRef]
- Cieslak, M.C.; Castelfranco, A.M.; Roncalli, V.; Lenz, P.H.; Hartline, D.K. t-Distributed Stochastic Neighbor Embedding (t-SNE): A tool for eco-physiological transcriptomic analysis. Mar. Genom. 2020, 51, 100723. [Google Scholar] [CrossRef]
- Graham, J.; Olchowski, A.; Gilreath, T. Review: A gentle introduction to imputation of missing values. Prev. Sci. 2007, 8, 206–213. [Google Scholar] [CrossRef]
- Lakshminarayan, K.; Harp, S.A.; Goldman, R.P.; Samad, T. Imputation of Missing Data Using Machine Learning Techniques. In KDD; AAAI Press: Portland, OR, USA, 1996. [Google Scholar]
- Raju, V.G.; Lakshmi, K.P.; Jain, V.M.; Kalidindi, A.; Padma, V. Study the influence of normalization/transformation process on the accuracy of supervised classification. In Proceedings of the 2020 Third International Conference on Smart Systems and Inventive Technology (ICSSIT), Tirunelveli, India, 20–22 August 2020. [Google Scholar]
- Singh, D.; Singh, B. Investigating the impact of data normalization on classification performance. Appl. Soft Comput. 2020, 97, 105524. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017, 30, 4765–4774. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. Consistent feature attribution for tree ensembles. arXiv 2017, arXiv:1706.06060. [Google Scholar]
- Abdel-Aal, R. GMDH-based feature ranking and selection for improved classification of medical data. J. Biomed. Inform. 2005, 38, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. Machine Learning, A Probabilistic Perspective; Taylor & Francis: Abingdon, UK, 2014. [Google Scholar]
- Rahman, T.; Khandakar, A.; Abir, F.F.; Faisal, M.A.A.; Hossain, M.S.; Podder, K.K.; Abbas, T.O.; Alam, M.F.; Kashem, S.B.; Islam, M.T. QCovSML: A reliable COVID-19 detection system using CBC biomarkers by a stacking machine learning model. Comput. Biol. Med. 2022, 143, 105284. [Google Scholar] [CrossRef] [PubMed]
- Keerthi, S.S.; Shevade, S.K.; Bhattacharyya, C.; Murthy, K.R.K. Improvements to Platt’s SMO algorithm for SVM classifier design. Neural Comput. 2001, 13, 637–649. [Google Scholar] [CrossRef]
- Sivasankari, S.; Surendiran, J.; Yuvaraj, N.; Ramkumar, M.; Ravi, C.; Vidhya, R. Classification of diabetes using multilayer perceptron. In Proceedings of the 2022 IEEE International Conference on Distributed Computing and Electrical Circuits and Electronics (ICDCECE), Ballari, India, 23–24 April 2022; pp. 1–5. [Google Scholar]
- Haque, F.; Reaz, M.B.; Chowdhury, M.E.; Shapiai, M.I.b.; Malik, R.A.; Alhatou, M.; Kobashi, S.; Ara, I.; Ali, S.H.; Bakar, A.A. A machine learning-based severity prediction tool for the Michigan neuropathy screening instrument. Diagnostics 2023, 13, 264. [Google Scholar] [CrossRef]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobot. 2013, 7, 21. [Google Scholar] [CrossRef]
- Faisal, M.A.A.; Chowdhury, M.E.; Khandakar, A.; Hossain, M.S.; Alhatou, M.; Mahmud, S.; Ara, I.; Sheikh, S.I.; Ahmed, M.U. An investigation to study the effects of Tai Chi on human gait dynamics using classical machine learning. Comput. Biol. Med. 2022, 142, 105184. [Google Scholar] [CrossRef]
- Al-Sarem, M.; Saeed, F.; Boulila, W.; Emara, A.H.; Al-Mohaimeed, M.; Errais, M. Feature selection and classification using CatBoost method for improving the performance of predicting Parkinson’s disease. In Proceedings of the Advances on Smart and Soft Computing: Proceedings of ICACIn, Casablanca, Morocco, 24–25 May 2020. [Google Scholar]
- Khandakar, A.; Chowdhury, M.E.; Reaz, M.B.I.; Ali, S.H.M.; Hasan, M.A.; Kiranyaz, S.; Rahman, T.; Alfkey, R.; Bakar, A.A.A.; Malik, R.A. A machine learning model for early detection of diabetic foot using thermogram images. Comput. Biol. Med. 2021, 137, 104838. [Google Scholar] [CrossRef]
- Choubey, D.K.; Kumar, M.; Shukla, V.; Tripathi, S.; Dhandhania, V.K. Comparative analysis of classification methods with PCA and LDA for diabetes. Curr. Diabetes Rev. 2020, 16, 833–850. [Google Scholar]
- Sharaff, A.; Gupta, H. Extra-tree classifier with metaheuristics approach for email classification. In Advances in Computer Communication and Computational Sciences: Proceedings of IC4S 2018; Springer: Berlin/Heidelberg, Germany, 2018; pp. 189–197. [Google Scholar]
- Pal, M. Random forest classifier for remote sensing classification. Int. J. Remote Sens. 2005, 26, 217–222. [Google Scholar] [CrossRef]
- Rokach, L. Ensemble methods for classifiers. In Data Mining and Knowledge Discovery Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 957–980. [Google Scholar]
- Opitz, D.; Maclin, R. Popular ensemble methods: An empirical study. J. Artif. Intell. Res. 1999, 11, 169–198. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Bennet, S.; Kaufmann, M.; Takami, K.; Sjaarda, C.; Douchant, K.; Moslinger, E.; Wong, H.; Reed, D.E.; Ellis, A.K.; Vanner, S. Small-molecule metabolome identifies potential therapeutic targets against COVID-19. Sci. Rep. 2022, 12, 10029. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Huang, X.; Fang, X.-Y.; Zheng, H.; Cai, S.-L.; Sun, A.-J.; Zhao, L.; Zhang, Y. Application of metabolomics in clinical and laboratory gastrointestinal oncology. World J. Gastrointest. Oncol. 2021, 13, 536. [Google Scholar] [CrossRef]
- Carr, V.R.; Chaguza, C. Metagenomics for surveillance of respiratory pathogens. Nat. Rev. Microbiol. 2021, 19, 285. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individual ML Model | Stacked ML Model (With Different Meta-classifiers) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Models | Acc | P | R | SP | F1 | Auc | Acc | P | R | SP | F1 | Auc |
| CatBoost | 94.58 | 94.58 | 94.58 | 94.59 | 94.58 | 98.57 | 95.83 | 95.83 | 95.83 | 95.82 | 95.83 | 98.78 |
| RandomForest | 94.16 | 94.16 | 94.16 | 94.14 | 94.16 | 97.74 | 96.25 | 96.25 | 96.25 | 96.27 | 96.25 | 98.21 |
| AdaBoost | 93.75 | 94.01 | 93.75 | 94.04 | 93.75 | 98.34 | 95.42 | 95.45 | 95.42 | 95.5 | 95.42 | 97.19 |
| ExtraTrees | 93.75 | 93.75 | 93.75 | 93.69 | 93.74 | 97.52 | 97.08 | 97.11 | 97.08 | 97.18 | 97.08 | 98.12 |
| LogisticRegression | 92.91 | 93 | 92.91 | 93.06 | 92.91 | 93.27 | 95 | 95 | 95 | 94.98 | 95 | 98.3 |
| ElasticNet | 92.91 | 93 | 92.91 | 93.06 | 92.91 | 94.73 | 95 | 95 | 95 | 94.98 | 95 | 98.3 |
| SVM | 92.5 | 92.5 | 92.5 | 92.47 | 92.5 | 93.54 | 95.83 | 95.83 | 95.83 | 95.82 | 95.83 | 98.28 |
| LDA | 91.25 | 91.24 | 91.25 | 91.18 | 91.24 | 96.23 | 95.42 | 95.45 | 95.42 | 95.5 | 95.42 | 97.99 |
| LGBM | 91.25 | 91.26 | 91.25 | 91.11 | 91.24 | 95.87 | 95.42 | 95.42 | 95.42 | 95.43 | 95.42 | 98.27 |
| MLPClassifier | 90.83 | 91.04 | 90.83 | 91.07 | 90.83 | 94.85 | 95 | 95 | 95 | 94.98 | 95 | 97.48 |
| XGBClassifier | 90.83 | 90.83 | 90.83 | 90.73 | 90.82 | 94.34 | 94.58 | 94.62 | 94.58 | 94.67 | 94.59 | 97.92 |
| Kneighbors | 89.58 | 89.61 | 89.58 | 89.65 | 89.58 | 94.55 | 94.58 | 94.59 | 94.58 | 94.6 | 94.58 | 96.45 |
| GradientBoost | 89.58 | 90.097 | 89.58 | 89.99 | 89.57 | 95.04 | 94.17 | 94.17 | 94.16 | 94.08 | 94.16 | 98.65 |
| Model | Sensitivity | Specificity | AUC | |
|---|---|---|---|---|
| Hogan et al. [28] | LGBM | 94% | 96% | 100% |
| Ours | Stacking-based Extra Trees | 100% | 100% | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumon, M.S.I.; Hossain, M.S.A.; Al-Sulaiti, H.; Yassine, H.M.; Chowdhury, M.E.H. Enhancing Influenza Detection through Integrative Machine Learning and Nasopharyngeal Metabolomic Profiling: A Comprehensive Study. Diagnostics 2024, 14, 2214. https://doi.org/10.3390/diagnostics14192214
Sumon MSI, Hossain MSA, Al-Sulaiti H, Yassine HM, Chowdhury MEH. Enhancing Influenza Detection through Integrative Machine Learning and Nasopharyngeal Metabolomic Profiling: A Comprehensive Study. Diagnostics. 2024; 14(19):2214. https://doi.org/10.3390/diagnostics14192214
Chicago/Turabian StyleSumon, Md. Shaheenur Islam, Md Sakib Abrar Hossain, Haya Al-Sulaiti, Hadi M. Yassine, and Muhammad E. H. Chowdhury. 2024. "Enhancing Influenza Detection through Integrative Machine Learning and Nasopharyngeal Metabolomic Profiling: A Comprehensive Study" Diagnostics 14, no. 19: 2214. https://doi.org/10.3390/diagnostics14192214
APA StyleSumon, M. S. I., Hossain, M. S. A., Al-Sulaiti, H., Yassine, H. M., & Chowdhury, M. E. H. (2024). Enhancing Influenza Detection through Integrative Machine Learning and Nasopharyngeal Metabolomic Profiling: A Comprehensive Study. Diagnostics, 14(19), 2214. https://doi.org/10.3390/diagnostics14192214