
STAG1 Disease, Central Precocious Puberty, and Bone Fragility—A Case Report

{kind=link}
Abstract
1. Introduction
2. Case Presentation
2.1. Clinical Findings
2.2. Diagnostic Assessment
2.3. Therapeutic Intervention and Follow-Up
3. Discussion
3.1. STAG1 Molecular and Functional Insights–“Typical” Clinical Features
3.2. CPP in This Case’s Context
- (1)
- (2)
- The neurodevelopmental abnormalities seen in STAG1-related disorders may indirectly influence hypothalamic function, leading to the early activation of puberty.
3.3. Bone Fragility in This Case’s Context
- (1)
- Defective osteoblast function or connective tissue involvement is due to the cohesin complex regulating genes involved in osteoblast differentiation and bone matrix formation, or to impaired DNA repair mechanisms.
- (2)
- Hormonal influence on bone health: The early activation of the HPG axis in CPP may lead to accelerated bone age. While early estrogen exposure initially increases bone mineralization, it also leads to premature epiphyseal closure, which may contribute to long-term bone fragility.
3.4. Challenges and Future Directions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ID | Intellectual disability |
| CPP | Central precocious puberty |
| DNA | Deoxyribonucleic acid |
| CdLS | Cornelia de Lange Syndrome |
| ADHD | Attention-deficit/hyperactivity disorder |
| LH | Luteinizing hormone |
| FSH | Follicle-stimulating hormone |
| MRI | Magnetic resonance imaging |
| EEG | Electroencephalography |
| MLPA | Multiplex Ligation-dependent Probe Amplification |
| NGS | Next-generation sequencing |
| WES | Whole-exome sequencing |
| HPG axis | Hypothalamic–pituitary–gonadal axis |
| GnRH | Gonadotropin-releasing hormone |
| BMI | Body Mass Index |
| WGS | Whole-genome sequencing |
| RNA sequencing | Ribonucleic acid sequencing |
References
- Barbero, J. Cohesins, cohesin-regulators: Role in Chromosome Segregation/Repair, Potential in Tumorigenesis. Atlas Genet. Cytogenet. Oncol. Haematol. 2012, 16, 157–164. [Google Scholar] [CrossRef]
- Cipriano, L.; Russo, R.; Andolfo, I.; Manno, M.; Piscopo, R.; Iolascon, A.; Piscopo, C. A Novel De Novo STAG1 Variant in Monozygotic Twins with Neurodevelopmental Disorder: New Insights in Clinical Heterogeneity. Genes 2024, 15, 1184. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.E.; Patel, V.; Jin, W.; Adam, M.P.; et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. 2019, 21, 663–675. [Google Scholar] [CrossRef]
- Di Muro, E.; Palumbo, P.; Benvenuto, M.; Accadia, M.; Di Giacomo, M.C.; Manieri, S.; Abate, R.; Tagliente, M.; Castellana, S.; Mazza, T.; et al. Novel STAG1 Frameshift Mutation in a Patient Affected by a Syndromic Form of Neurodevelopmental Disorder. Genes 2021, 12, 1116. [Google Scholar] [CrossRef]
- Funato, M.; Uehara, T.; Okada, Y.; Kaneko, H.; Kosaki, K. Cohesinopathy presenting with microtia, facial palsy, and hearing loss caused by STAG1 pathogenic variant. Congenit. Anom. 2022, 62, 82–83. [Google Scholar] [CrossRef]
- Lehalle, D.; Mosca-Boidron, A.L.; Begtrup, A.; Boute-Benejean, O.; Charles, P.; Cho, M.T.; Clarkson, A.; Devinsky, O.; Duffourd, Y.; Duplomb-Jego, L.; et al. STAG1 mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J. Med. Genet. 2017, 54, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Labudina, A.A.; Meier, M.; Gimenez, G.; Tatarakis, D.; Ketharnathan, S.; Mackie, B.; Schilling, T.F.; Antony, J.; Horsfield, J.A. Cohesin composition and dosage independently affect early development in zebrafish. Development 2024, 151, dev202593. [Google Scholar] [CrossRef]
- Avagliano, L.; Parenti, I.; Grazioli, P.; Di Fede, E.; Parodi, C.; Mariani, M.; Kaiser, F.J.; Selicorni, A.; Gervasini, C.; Massa, V. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin. Genet. 2020, 97, 3–11. [Google Scholar] [CrossRef]
- Deschamps, G.N. Cornelia de Lange Syndrome. Neonatal Netw. 2022, 41, 145–149. [Google Scholar] [CrossRef]
- Bregvadze, K.; Sukhiashvili, A.; Lartsuliani, M.; Melikidze, E.; Tkemaladze, T. A novel STAG1 variant associated with congenital clubfoot and microphthalmia: A case report. SAGE Open Med. Case Rep. 2024, 12, 2050313X241277123. [Google Scholar] [CrossRef]
- Canton, A.P.M.; Seraphim, C.E.; Montenegro, L.R.; Krepischi, A.C.V.; Mendonca, B.B.; Latronico, A.C.; Brito, V.N. The genetic etiology is a relevant cause of central precocious puberty. Eur. J. Endocrinol. 2024, 190, 479–488. [Google Scholar] [CrossRef]
- Moise-Silverman, J.; Silverman, L.A. A review of the genetics and epigenetics of central precocious puberty. Front. Endocrinol. 2022, 13, 1029137. [Google Scholar] [CrossRef] [PubMed]
- Brito, V.N.; Canton, A.P.; Seraphim, C.E.; Abreu, A.P.; Macedo, D.B.; Mendonca, B.B.; Kaiser, U.B.; Argente, J.; Latronico, A.C. The congenital and acquired mechanisms implicated in the etiology of central precocious puberty. Endocr. Rev. 2023, 44, 193–221. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M. Part I: Which child with a chronic disease needs bone health monitoring? Curr. Osteoporos. Rep. 2021, 19, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Charoenngam, N.; Cevik, M.B.; Holick, M.F. Diagnosis and management of pediatric metabolic bone diseases associated with skeletal fragility. Curr. Opin. Pediatr. 2020, 32, 560–573. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Roberts, S.A.; Kaiser, U.B. GENETICS IN ENDOCRINOLOGY: Genetic etiologies of central precocious puberty and the role of imprinted genes. Eur. J. Endocrinol. 2020, 183, R107–R117. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, N.; Pati, D. Cohesin subunit RAD21: From biology to disease. Gene 2020, 758, 144966. [Google Scholar] [CrossRef]
- Zhao, B.; Lin, J.; Rong, L.; Wu, S.; Deng, Z.; Fatkhutdinov, N.; Zundell, J.; Fukumoto, T.; Liu, Q.; Kossenkov, A.; et al. ARID1A promotes genomic stability through protecting telomere cohesion. Nat. Commun. 2019, 10, 4067. [Google Scholar] [CrossRef]
- Soldin, O.P.; Hoffman, E.G.; Waring, M.A.; Soldin, S.J. Pediatric reference intervals for FSH, LH, estradiol, T3, free T3, cortisol, and growth hormone on the DPC IMMULITE 1000. Clin. Chim. Acta 2005, 355, 205–210. [Google Scholar] [CrossRef]
- Bangalore Krishna, K.; Garibaldi, L. Critical appraisal of diagnostic laboratory tests in the evaluation of central precocious puberty. Front. Pediatr. 2025, 12, 1504874. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, I.S.C.; Çelik, N.; Bolat, S. Evaluation of Serum Adipokine Levels in Girls With Central Precocious Puberty. Clin. Endocrinol. 2024, 102, 413–420. [Google Scholar] [CrossRef]
- Çatlı, G.; Erdem, P.; Anık, A.; Abacı, A.; Böber, E. Clinical and laboratory findings in the differential diagnosis of central precocious puberty and premature thelarche. Turk. Arch. Pediatr./Türk Pediatri Arşivi 2015, 50, 20. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Alghamdi, A.H. Precocious puberty: Types, pathogenesis and updated management. Cureus 2023, 15, e47485. [Google Scholar] [CrossRef]
- Zhao, H.-Y.; Zhang, Y.-R.; Zhang, R.; Li, Y.-T.; Guo, R.-L.; Shi, W.-S. Comprehensive analysis of untargeted metabolomics and lipidomics in girls with central precocious puberty. Front. Pediatr. 2023, 11, 1157272. [Google Scholar] [CrossRef] [PubMed]
- Ketharnathan, S.; Labudina, A.; Horsfield, J.A. Cohesin Components Stag1 and Stag2 Differentially Influence Haematopoietic Mesoderm Development in Zebrafish Embryos. Front. Cell Dev. Biol. 2020, 8, 617545. [Google Scholar] [CrossRef]
- Viny, A.D.; Bowman, R.L.; Liu, Y.; Lavallée, V.P.; Eisman, S.E.; Xiao, W.; Durham, B.H.; Navitski, A.; Park, J.; Braunstein, S.; et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 2019, 25, 682–696.e688. [Google Scholar] [CrossRef]
- Arruda, N.L.; Carico, Z.M.; Justice, M.; Liu, Y.F.; Zhou, J.; Stefan, H.C.; Dowen, J.M. Distinct and overlapping roles of STAG1 and STAG2 in cohesin localization and gene expression in embryonic stem cells. Epigenetics Chromatin 2020, 13, 32. [Google Scholar] [CrossRef]
- van der Lelij, P.; Lieb, S.; Jude, J.; Wutz, G.; Santos, C.P.; Falkenberg, K.; Schlattl, A.; Ban, J.; Schwentner, R.; Hoffmann, T.; et al. Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. Elife 2017, 6, e26980. [Google Scholar] [CrossRef]
- Saitta, C.; Rebellato, S.; Bettini, L.R.; Giudici, G.; Panini, N.; Erba, E.; Massa, V.; Auer, F.; Friedrich, U.; Hauer, J.; et al. Potential role of STAG1 mutations in genetic predisposition to childhood hematological malignancies. Blood Cancer J. 2022, 12, 88. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Signer, R.H.; Center, U.C.G.; Vilain, E.; Martinez-Agosto, J.A. Mutations in STAG2 cause an X-linked cohesinopathy associated with undergrowth, developmental delay, and dysmorphia: Expanding the phenotype in males. Mol. Genet. Genom. Med. 2019, 7, e00501. [Google Scholar] [CrossRef] [PubMed]
- Yingjun, X.; Wen, T.; Yujian, L.; Lingling, X.; Huimin, H.; Qun, F.; Junhong, C. Microduplication of chromosome Xq25 encompassing STAG2 gene in a boy with intellectual disability. Eur. J. Med. Genet. 2015, 58, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Della Giustina, E.; Salviato, T.; Caramaschi, S.; Fabbiani, L.; Reggiani Bonetti, L. Cornelia de Lange Syndrome: Expanding the Neuropathological Spectrum and Clinical Correlations. Fetal Pediatr. Pathol. 2024, 43, 477–486. [Google Scholar] [CrossRef]
- Canton, A.P.M.; Macedo, D.B.; Abreu, A.P.; Latronico, A.C. Genetics and Epigenetics of Human Pubertal Timing: The Contribution of Genes Associated With Central Precocious Puberty. J. Endocr. Soc. 2025, 9, bvae228. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A. Genetic and epigenetic aspects of the KISS1 and KISS1R genes in pubertal development and central precocious puberty: A review. Precis. Future Med. 2023, 7, 137–146. [Google Scholar] [CrossRef]
- Malay, J.; George, B.T.; Dube, R.; Kar, S.S.; Rangraze, I.R. Exploring the Rise in Precocious Puberty: Interplay of Genetic and Environmental Factors. Preprints 2024. [Google Scholar] [CrossRef]
- Gu, X.; Xiong, W.; Yang, Y.; Li, H.; Xiong, C. A comprehensive meta-analysis to identify susceptibility genetic variants for precocious puberty. Ann. Hum. Genet. 2024, 88, 138–153. [Google Scholar] [CrossRef]
- Narusawa, H.; Ogawa, T.; Yagasaki, H.; Nagasaki, K.; Urakawa, T.; Saito, T.; Soneda, S.; Kinjo, S.; Sano, S.; Mamada, M. Comprehensive study on central precocious puberty: Molecular and clinical analyses in 90 patients. J. Clin. Endocrinol. Metab. 2024, 110, dgae666. [Google Scholar] [CrossRef]
- Heddar, A.; Dessen, P.; Flatters, D.; Misrahi, M. Novel STAG3 mutations in a Caucasian family with primary ovarian insufficiency. Mol. Genet. Genom. 2019, 294, 1527–1534. [Google Scholar] [CrossRef]
- Seymour, H.; Feben, C.; Nevondwe, P.; Kerr, R.; Spencer, C.; Mudau, M.; Honey, E.; Lombard, Z.; Krause, A.; Carstens, N. Mutation profiling in South African patients with Cornelia de Lange syndrome phenotype. Mol. Genet. Genom. Med. 2024, 12, e2342. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Bando, M.; Itoh, T.; Deardorff, M.A.; Clark, D.; Kaur, M.; Tandy, S.; Kondoh, T.; Rappaport, E.; et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009, 7, e1000119. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gil, D.; Losada, A. NIPBL and cohesin: New take on a classic tale. Trends Cell Biol. 2023, 33, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Maione, L.; Bouvattier, C.; Kaiser, U.B. Central precocious puberty: Recent advances in understanding the aetiology and in the clinical approach. Clin. Endocrinol. 2021, 95, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Giusti, F.; Iantomasi, T.; Brandi, M.L. Congenital metabolic bone disorders as a cause of bone fragility. Int. J. Mol. Sci. 2021, 22, 10281. [Google Scholar] [CrossRef]
- Testa, E.J.; Callanan, T.C.; Evans, A.R.; Aaron, R.K. Osteoporosis and fragility fractures. Rhode Isl. Med. J. 2022, 105, 15–21. [Google Scholar]
- Singh, V.; Pal, A.K.; Biswas, D.; Ghosh, A.; Singh, B.P. Evaluation of environmental and socioeconomic factors contributing to fragility fractures in Indians. medRxiv 2020. [Google Scholar] [CrossRef]
 indicates who the proband is.
indicates who the proband is.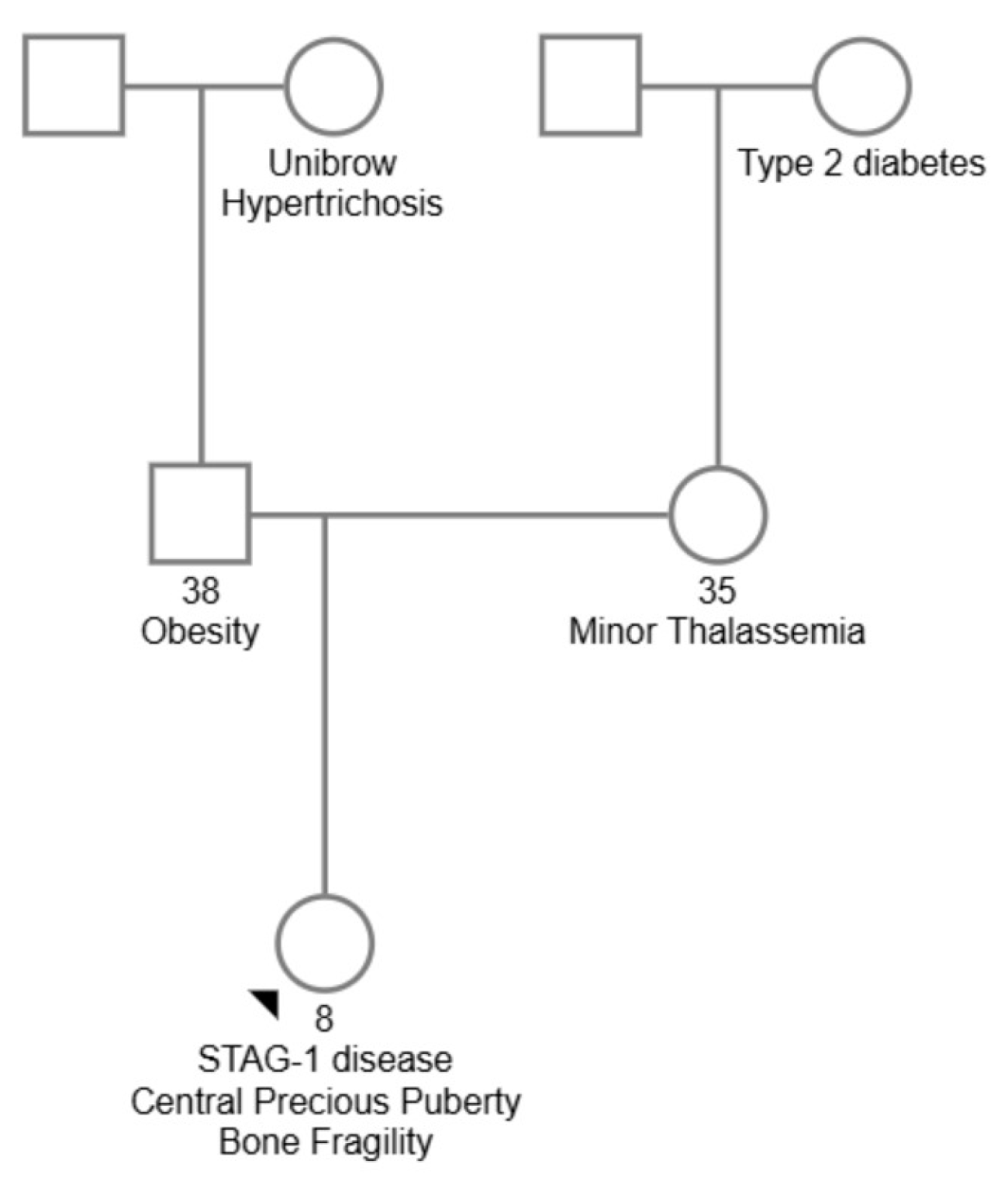
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Șerban, R.-C.; Mituț-Velișcu, A.-M.; Costache, A.; Cima, L.-N.; Niculescu, C.; Moroșanu, A.; Riza, A.-L.; Streață, I. STAG1 Disease, Central Precocious Puberty, and Bone Fragility—A Case Report. Diagnostics 2025, 15, 1076. https://doi.org/10.3390/diagnostics15091076
Șerban R-C, Mituț-Velișcu A-M, Costache A, Cima L-N, Niculescu C, Moroșanu A, Riza A-L, Streață I. STAG1 Disease, Central Precocious Puberty, and Bone Fragility—A Case Report. Diagnostics. 2025; 15(9):1076. https://doi.org/10.3390/diagnostics15091076
Chicago/Turabian StyleȘerban, Rebecca-Cristiana, Andreea-Mădălina Mituț-Velișcu, Andrei Costache, Luminița-Nicoleta Cima, Carmen Niculescu, Aritina Moroșanu, Anca-Lelia Riza, and Ioana Streață. 2025. "STAG1 Disease, Central Precocious Puberty, and Bone Fragility—A Case Report" Diagnostics 15, no. 9: 1076. https://doi.org/10.3390/diagnostics15091076
APA StyleȘerban, R.-C., Mituț-Velișcu, A.-M., Costache, A., Cima, L.-N., Niculescu, C., Moroșanu, A., Riza, A.-L., & Streață, I. (2025). STAG1 Disease, Central Precocious Puberty, and Bone Fragility—A Case Report. Diagnostics, 15(9), 1076. https://doi.org/10.3390/diagnostics15091076