
Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure
2.1. HMGB1
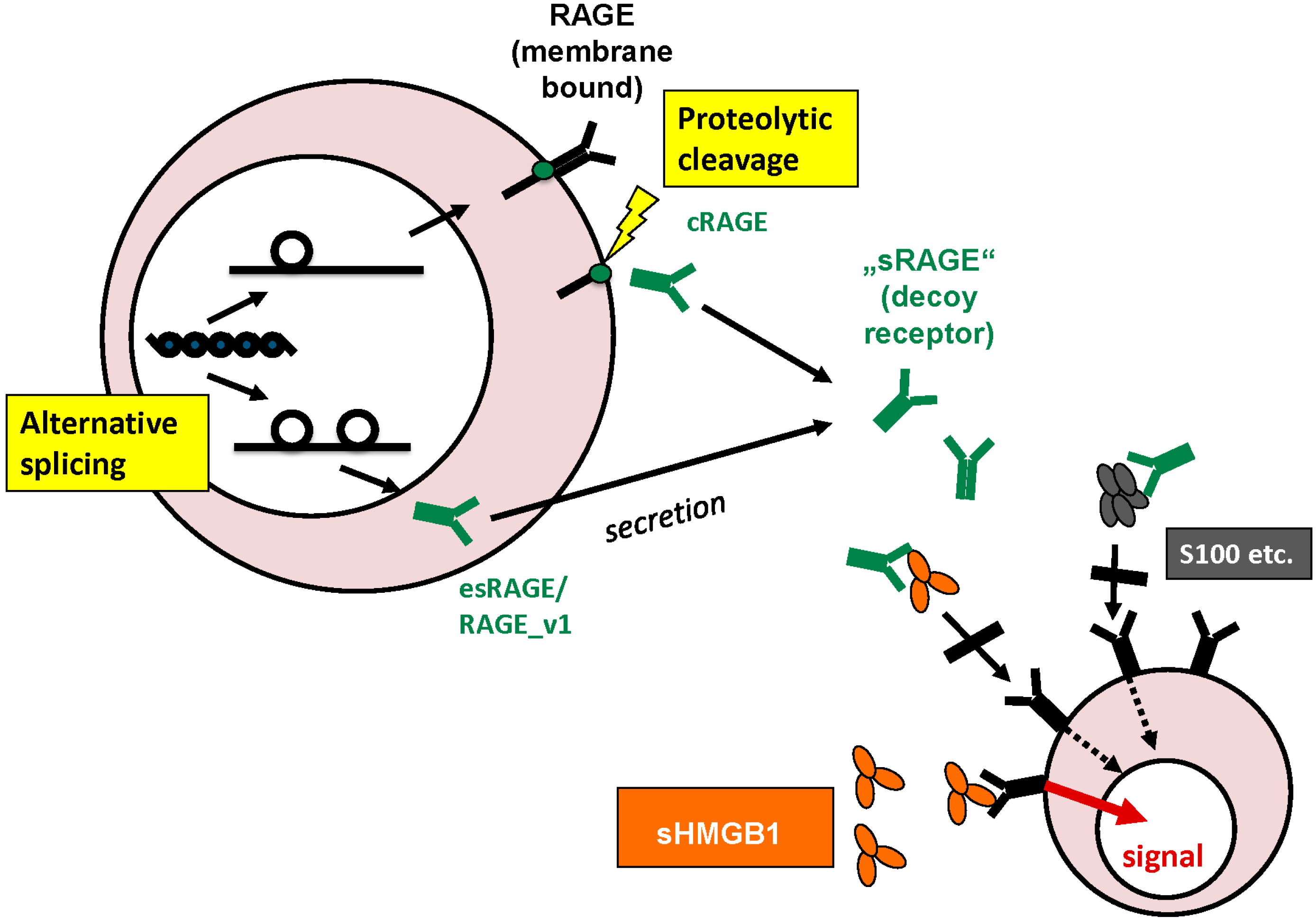
2.2. RAGE

3. Physiological Functions and Cellular Release
3.1. Intracellular Functions of HMGB1
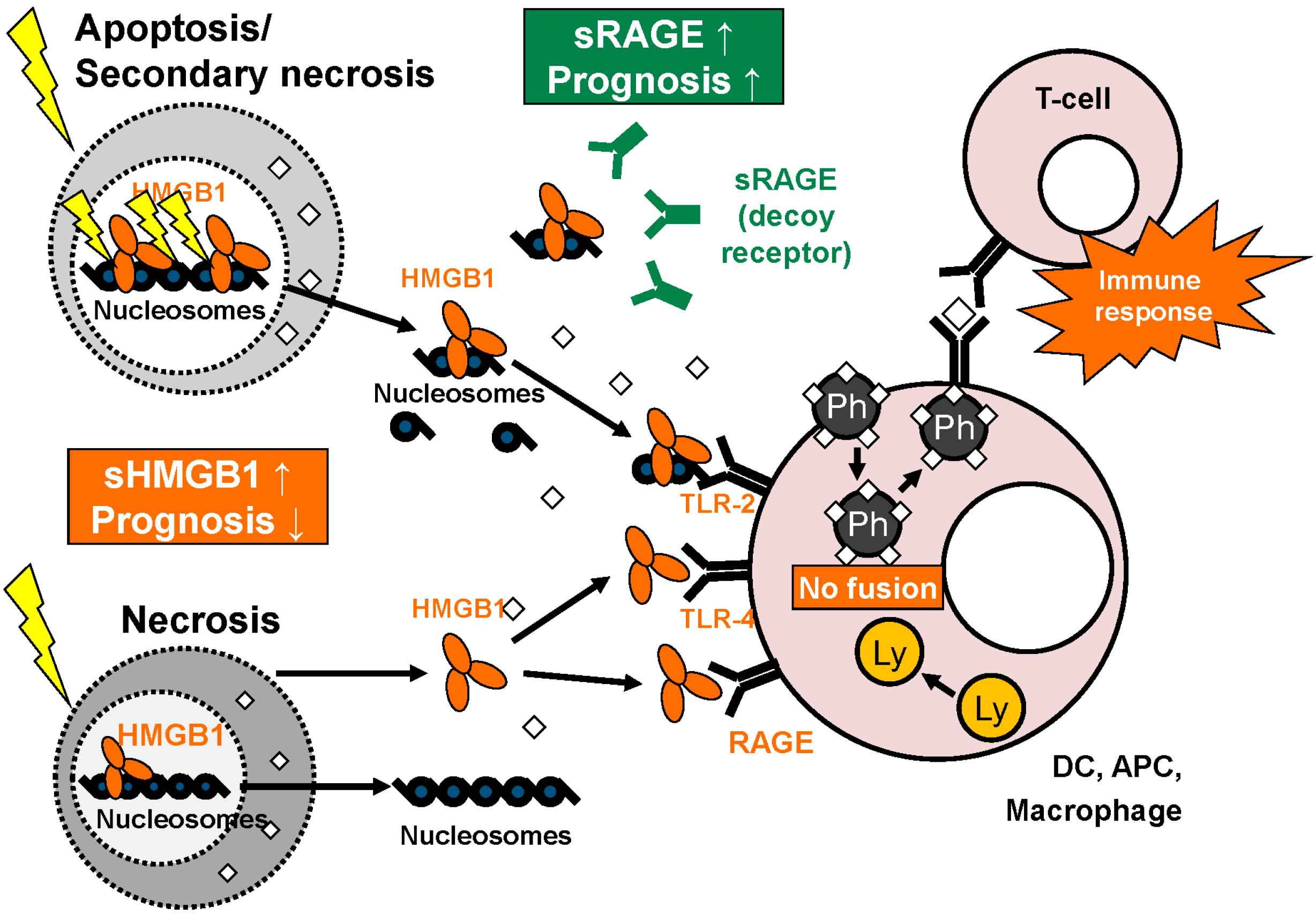
3.2. Cellular Release of HMGB1
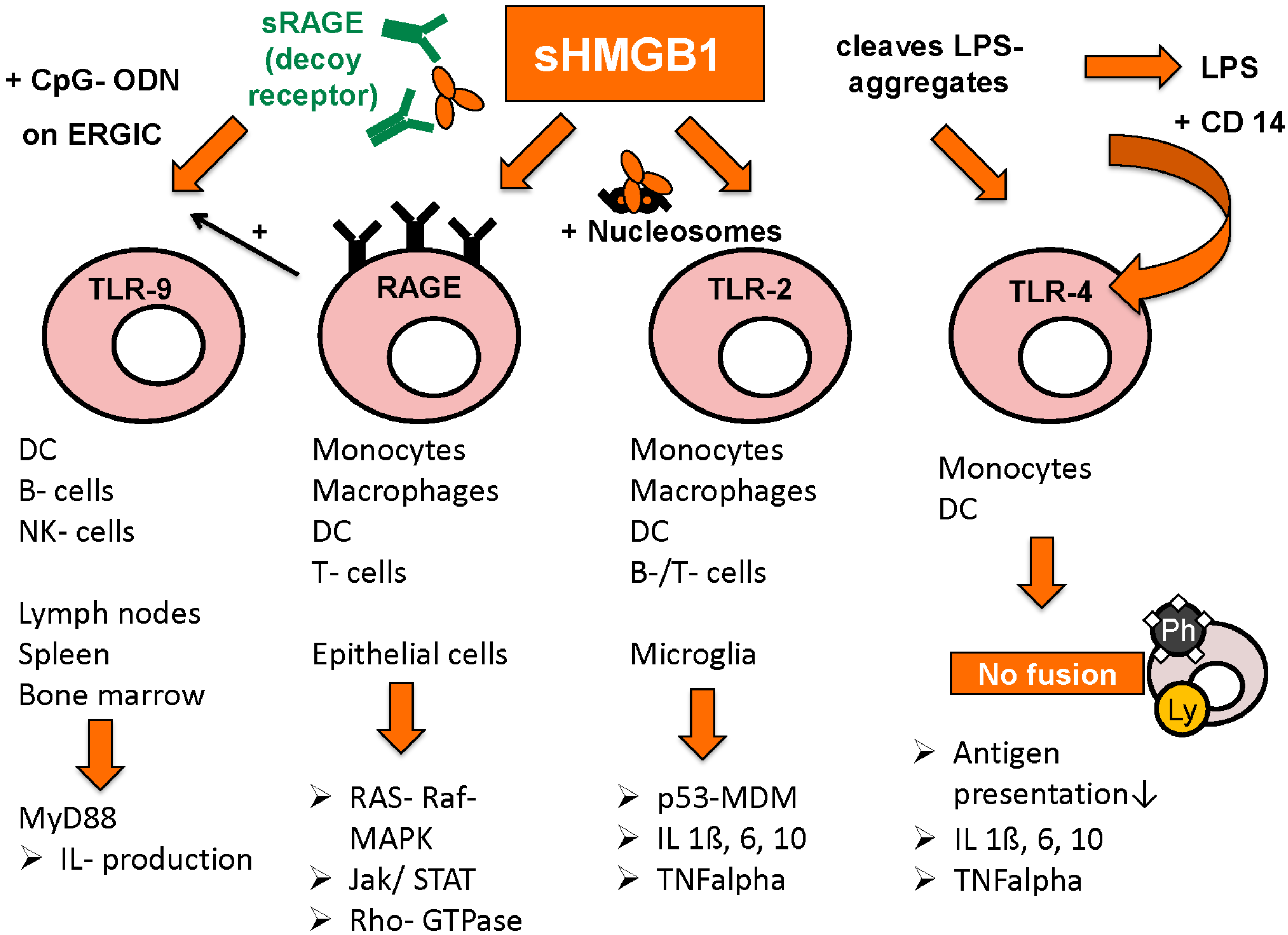
3.3. Extracellular Functions of HMGB1


3.4. Intracellular and Extracellular Functions of RAGE
4. Pathophysiological Functions
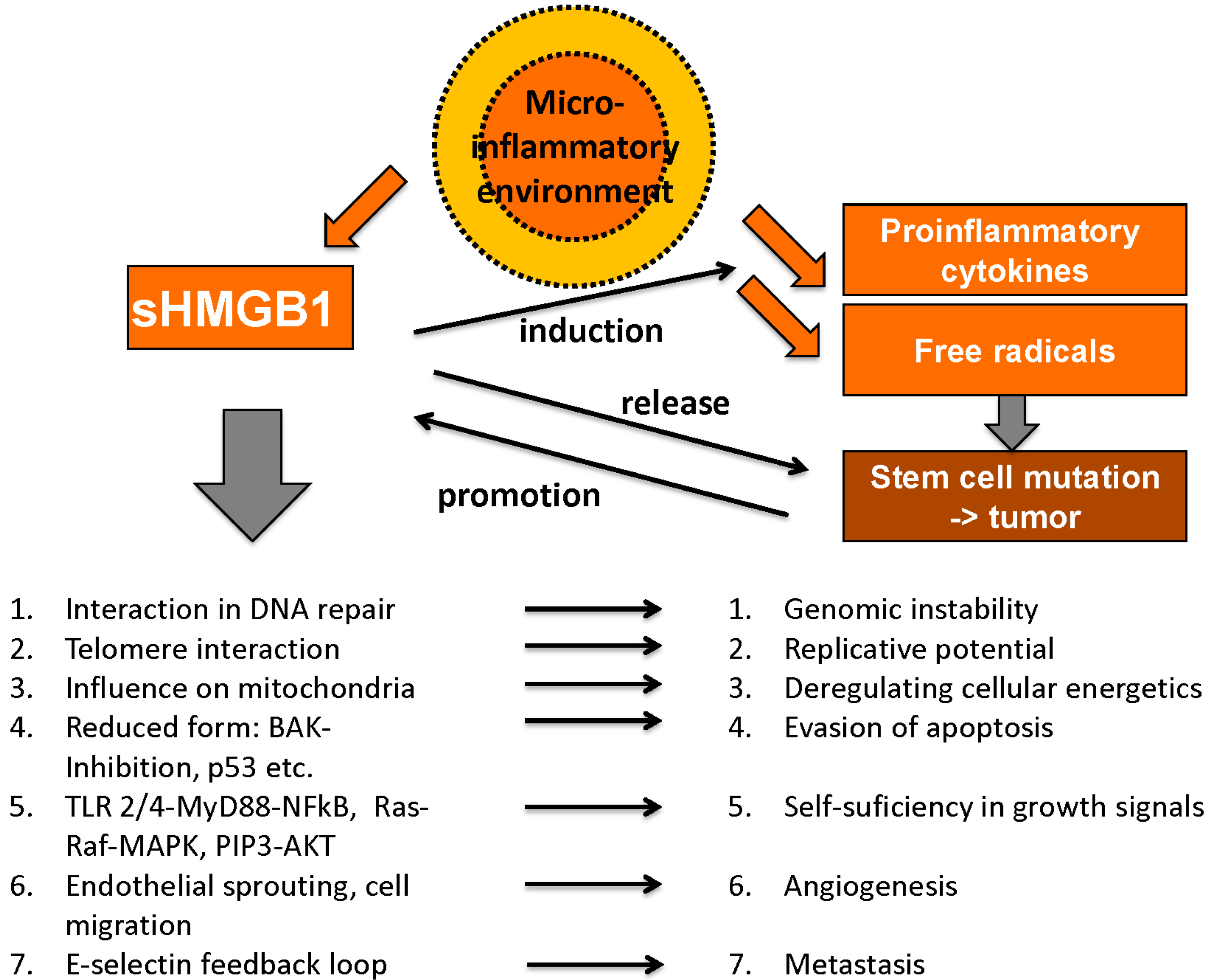
4.1. Role of HMGB1 and RAGE in Malignant Disease

4.1.1. Unlimited Replicative Potential
4.1.2. Genome Instability and Mutation
4.1.3. Evasion of Apoptosis
4.1.4. Self-Sufficiency in Growth Signals
4.1.5. Insensitivity to Inhibitors of Growth
4.1.6. Deregulating Cellular Energetics
4.1.7. Angiogenesis
4.1.8. Metastasis and Tissue Invasion
4.1.9. Microinflammatory Environment
Avoiding Immune Destruction
4.2. Role of HMGB1 and RAGE in Autoimmune Disease and Non-Malignant Diseases
4.2.1. Systemic Lupus Erythematosus
4.2.2. Rheumatoid Arthritis
4.2.3. Acute Inflammation
5. Methods for Detection of HMGB1 and sRAGE
6. Clinical Relevance of Circulating HMGB1 and sRAGE
6.1. Clinical Relevance of Circulating HMGB1 and sRAGE in Autoimmune Disease and Non-Malignant Disease
6.2. Clinical Relevance of Circulating HMGB1 and sRAGE in Malignant Disease
7. Conclusions and Perspectives
Conflicts of Interest
References
- Stieber, P.; Heinemann, V. Sinnvoller einsatz von tumormarkern/Sensible use of tumor markers. Laboratoriumsmedizin 2008, 32, 339–360. [Google Scholar] [CrossRef]
- Sturgeon, C.M.; Duffy, M.J.; Hofmann, B.R.; Lamerz, R.; Fritsche, H.A.; Gaarenstroom, K.; Bonfrer, J.; Ecke, T.H.; Grossman, H.B.; Hayes, P.; et al. National academy of clinical biochemistry laboratory medicine practice guidelines for use of tumor markers in liver, bladder, cervical, and gastric cancers. Clin. Chem. 2010, 56, e1–e48. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008, 15, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Lotze, M.T.; Latz, E.; Manfredi, A. Mechanisms of sterile inflammation. Front. Immunol. 2013, 4, 398. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Soluble form of a receptor for advanced glycation end products (sRAGE) as a biomarker. Front. Biosci. 2010, 2, 1184–1195. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. Hmgb1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the united states, national cancer institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Brambs, H.J.; Claussen, C.D. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993, 25, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Gress, F.; Gottlieb, K.; Sherman, S.; Lehman, G. Endoscopic ultrasonography-guided fine-needle aspiration biopsy of suspected pancreatic cancer. Ann. Intern. Med. 2001, 134, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Westwood, M.; Joore, M.; Whiting, P.; van Asselt, T.; Ramaekers, B.; Armstrong, N.; Misso, K.; Severens, J.; Kleijnen, J. Epidermal growth factor receptor tyrosine kinase (EGFR-TK) mutation testing in adults with locally advanced or metastatic non-small cell lung cancer: A systematic review and cost-effectiveness analysis. Health Technol. Assess. 2014, 18, 1–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboue, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Stieber, P.; Holdenrieder, S. Lung cancer biomarkers—Where we are and what we need. Cancer Biomarks 2010, 6, 221–224. [Google Scholar]
- Behesnilian, A.S.; Reiter, R.E. Risk stratification of prostate cancer in the modern era. Curr. Opin. Urol. 2015, 25, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Heinemann, V.; Kullmann, F.; Laubender, R.P.; Klose, C.; Bruns, C.J.; Holdenrieder, S.; Modest, D.P.; Schulz, C.; Boeck, S. Prognostic value of CA 19-9, CEA, CRP, LDH and bilirubin levels in locally advanced and metastatic pancreatic cancer: Results from a multicenter, pooled analysis of patients receiving palliative chemotherapy. J. Cancer Res. Clin. Oncol. 2013, 139, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, C.; Hill, R.; Hortin, G.L.; Thompson, D. Taking a new biomarker into routine use—A perspective from the routine clinical biochemistry laboratory. Proteomics. Clin. Appl. 2010, 4, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Holdenrieder, S.; Zhi, X.Y.; Zhang, X.; Qiu, L.; Geistanger, A.; Lisy, M.R.; Wehnl, B.; van den Broek, D.; Escudero, J.M.; et al. Multicenter evaluation of a new progastrin-releasing peptide (ProGRP) immunoassay across Europe and China. Clin. Chim. Acta 2015, 438, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Molina, R.; Escudero, J.M.; Auge, J.M.; Filella, X.; Foj, L.; Torne, A.; Lejarcegui, J.; Pahisa, J. HE4 a novel tumour marker for ovarian cancer: Comparison with CA 125 and ROMA algorithm in patients with gynaecological diseases. Tumour Biol. 2011, 32, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Zamanian-Azodi, M.; Rezaei-Tavirani, M.; Hasanzadeh, H.; Rahmati Rad, S.; Dalilan, S. Introducing biomarker panel in esophageal, gastric, and colon cancers; a proteomic approach. Gastroenterol. Hepatol. Bed Bench 2015, 8, 6–18. [Google Scholar] [PubMed]
- Goodwin, G.H.; Johns, E.W. The isolation and purification of the high mobility group (HMG) nonhistone chromosomal proteins. Methods Cell Biol. 1977, 16, 257–267. [Google Scholar] [PubMed]
- Bustin, M.; Lehn, D.A.; Landsman, D. Structural features of the hmg chromosomal proteins and their genes. Biochim. Biophys. Acta 1990, 1049, 231–243. [Google Scholar] [CrossRef]
- Saito, K.; Kikuchi, T.; Yoshida, M. The mechanism of sequence non-specific DNA binding of HMG1/2-box B in HMG1 with DNA. Protein Eng. 1999, 12, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Chou, H.; Kawase, T.; Shirakawa, H.; Yoshida, M. Acidic C-tail of HMGB1 is required for its target binding to nucleosome linker DNA and transcription stimulation. Biochemistry 2004, 43, 9901–9908. [Google Scholar] [CrossRef] [PubMed]
- Bustin, M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell Biol. 1999, 19, 5237–5246. [Google Scholar] [PubMed]
- Ohndorf, U.M.; Rould, M.A.; He, Q.; Pabo, C.O.; Lippard, S.J. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature 1999, 399, 708–712. [Google Scholar] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Finelli, P.; Rocchi, M.; Bianchi, M.E. The active gene that encodes human high mobility group 1 protein (HMG1) contains introns and maps to chromosome 13. Genomics 1996, 35, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Kornblit, B.; Munthe-Fog, L.; Petersen, S.L.; Madsen, H.O.; Vindelov, L.; Garred, P. The genetic variation of the human HMGB1 gene. Tissue Antigens 2007, 70, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Stokrova, J.; Thomas, J.O. DNA looping by the HMG-box domains of HMG1 and modulation of DNA binding by the acidic C-terminal domain. Nucl. Acids Res. 1994, 22, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Izumi, H.; Nagatani, G.; Ohmori, H.; Nagasue, N.; Ise, T.; Yoshida, T.; Yasumoto, K.; Kohno, K. Physical interaction of tumour suppressor p53/p73 with CCAAT-binding transcription factor 2 (CTF2) and differential regulation of human high-mobility group 1 (HMG1) gene expression. Biochem. J. 2003, 371, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Rothermund, K.; Rogulski, K.; Fernandes, E.; Whiting, A.; Sedivy, J.; Pu, L.; Prochownik, E.V. C-Myc-independent restoration of multiple phenotypes by two C-Myc target genes with overlapping functions. Cancer Res. 2005, 65, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Pogna, E.A.; Clayton, A.L.; Mahadevan, L.C. Signalling to chromatin through post-translational modifications of HMGN. Biochim. Biophys. Acta 2010, 1799, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Fukazawa, J.; Yoshida, M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 2007, 282, 16336–16344. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.K.; Chao, C.C. The cytokine activity of HMGB1—Extracellular escape of the nuclear protein. Chang Gung Med. J. 2005, 28, 673–682. [Google Scholar] [PubMed]
- Muller, S.; Ronfani, L.; Bianchi, M.E. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J. Intern. Med. 2004, 255, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Mosevitsky, M.I.; Novitskaya, V.A.; Iogannsen, M.G.; Zabezhinsky, M.A. Tissue specificity of nucleo-cytoplasmic distribution of HMG1 and HMG2 proteins and their probable functions. Eur. J. Biochem. 1989, 185, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Seyedin, S.M.; Pehrson, J.R.; Cole, R.D. Loss of chromosomal high mobility group proteins HMG1 and HMG2 when mouse neuroblastoma and friend erythroleukemia cells become committed to differentiation. Proc. Natl. Acad. Sci. USA 1981, 78, 5988–5992. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Kawakami, Y.; Arai, T.; Imuta, H.; Fujiwara, M.; Kanma, H.; Yamashita, K. Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem. Biophys. Res. Commun. 1997, 238, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Demling, N.; Ehrhardt, C.; Kasper, M.; Laue, M.; Knels, L.; Rieber, E.P. Promotion of cell adherence and spreading: A novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type i cells. Cell Tissue Res. 2006, 323, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar] [PubMed]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. Rage drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via rage and its ligands in apoE-/- mice. J. Clin. Invest. 2008, 118, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Ritthaler, U.; Deng, Y.; Zhang, Y.; Greten, J.; Abel, M.; Sido, B.; Allenberg, J.; Otto, G.; Roth, H.; Bierhaus, A.; et al. Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am. J. Pathol. 1995, 146, 688–694. [Google Scholar] [PubMed]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [PubMed]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar] [PubMed]
- Malherbe, P.; Richards, J.G.; Gaillard, H.; Thompson, A.; Diener, C.; Schuler, A.; Huber, G. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and swedish mutant amyloid precursor protein. Mol. Brain Res. 1999, 71, 159–170. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. Rage is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Du Yan, S.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. Nε-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [PubMed]
- Dattilo, B.M.; Fritz, G.; Leclerc, E.; Kooi, C.W.; Heizmann, C.W.; Chazin, W.J. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry 2007, 46, 6957–6970. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bukulin, M.; Kojro, E.; Roth, A.; Metz, V.V.; Fahrenholz, F.; Nawroth, P.P.; Bierhaus, A.; Postina, R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 2008, 283, 35507–35516. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of rage gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Schmidt, A.M.; Hudson, B.I. Alternative splicing of rage: Roles in biology and disease. Front. Biosci. 2011, 16, 2756–2770. [Google Scholar] [CrossRef]
- Geroldi, D.; Falcone, C.; Emanuele, E. Soluble receptor for advanced glycation end products: From disease marker to potential therapeutic target. Curr. Med. Chem. 2006, 13, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Stros, M. Hmgb proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta 2010, 1799, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Polanska, E.; Struncova, S.; Pospisilova, S. HMGB1 and HMGB2 proteins up-regulate cellular expression of human topoisomerase IIalpha. Nucl. Acids Res. 2009, 37, 2070–2086. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T.; Langst, G.; Strohner, R.; Becker, P.B.; Bianchi, M.E. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 2002, 21, 6865–6873. [Google Scholar] [CrossRef] [PubMed]
- Sutrias-Grau, M.; Bianchi, M.E.; Bernues, J. High mobility group protein 1 interacts specifically with the core domain of human TATA box-binding protein and interferes with transcription factor IIB within the pre-initiation complex. J. Biol. Chem. 1999, 274, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, L.; Moorthy, N.C.; Murthy, K.G.; Manley, J.L.; Bustin, M.; Prives, C. High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev. 1998, 12, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Ozaki, T.; Bacikova, A.; Kageyama, H.; Nakagawara, A. HMGB1 and HMGB2 cell-specifically down-regulate the p53- and p73-dependent sequence-specific transactivation from the human Bax gene promoter. J. Biol. Chem. 2002, 277, 7157–7164. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, H.; Tang, Y.; Fan, Z.; Xiao, X.; Chen, F. High-mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF-κB and Egr-1. Thromb. Haemost. 2009, 102, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Agresti, A.; Lupo, R.; Bianchi, M.E.; Muller, S. Hmgb1 interacts differentially with members of the Rel family of transcription factors. Biochem. Biophys. Res. Commun. 2003, 302, 421–426. [Google Scholar] [CrossRef]
- Zappavigna, V.; Falciola, L.; Helmer-Citterich, M.; Mavilio, F.; Bianchi, M.E. HMG1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J. 1996, 15, 4981–4991. [Google Scholar] [PubMed]
- Jiao, Y.; Wang, H.C.; Fan, S.J. Growth suppression and radiosensitivity increase by HMGB1 in breast cancer. Acta Pharmacol. Sin. 2007, 28, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, D.C.; Hiom, K.; Paull, T.T.; Gellert, M. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 1997, 16, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Christensen, J.; Tattersall, P. Two widely spaced initiator binding sites create an HMG1-dependent parvovirus rolling-hairpin replication origin. J. Virol. 2000, 74, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Topalova, D.; Ugrinova, I.; Pashev, I.G.; Pasheva, E.A. HMGB1 protein inhibits DNA replication in vitro: A role of the acetylation and the acidic tail. Int. J. Biochem. Cell Biol. 2008, 40, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Cherny, D.; Jovin, T.M. HMG1 protein stimulates DNA end joining by promoting association of DNA molecules via their ends. Eur. J. Biochem. 2000, 267, 4088–4097. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Mitchell, D.L.; Vasquez, K.M. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 10320–10325. [Google Scholar] [CrossRef] [PubMed]
- Moggs, J.G.; Szymkowski, D.E.; Yamada, M.; Karran, P.; Wood, R.D. Differential human nucleotide excision repair of paired and mispaired cisplatin-DNA adducts. Nucl. Acids Res. 1997, 25, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Prasad, R.; Wilson, S.H. Hmgb1: Roles in base excision repair and related function. Biochim. Biophys. Acta 2010, 1799, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.C.; Christensen, J.; Vasquez, K.M. Interplay between human high mobility group protein 1 and replication protein a on psoralen-cross-linked DNA. Biochemistry 2005, 44, 4188–4195. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Reddy, M.C.; Vasquez, K.M. Human HMGB1 directly facilitates interactions between nucleotide excision repair proteins on triplex-directed psoralen interstrand crosslinks. DNA Repair 2009, 8, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Hou, W.; Yan, Z.; Chen, R.; Bonaroti, J.; Bansal, P.; Billiar, T.R.; Tsung, A.; Wang, Q.; et al. Intracellular HMGB1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014, 146, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; van Houten, B.; Zeh, H.J.; Billiar, T.R.; Lotze, M.T. High mobility group box 1 (HMGB1) phenotypic role revealed with stress. Mol. Med. 2014, 20, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Messer, J.S.; Wang, Y.; Lin, F.; Cham, C.M.; Chang, J.; Billiar, T.R.; Lotze, M.T.; Boone, D.L.; Chang, E.B. Cytosolic hmgb1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Invest. 2015, 125, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Ditsworth, D.; Zong, W.X.; Thompson, C.B. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 2007, 282, 17845–17854. [Google Scholar] [CrossRef] [PubMed]
- Kamau, E.; Takhampunya, R.; Li, T.; Kelly, E.; Peachman, K.K.; Lynch, J.A.; Sun, P.; Palmer, D.R. Dengue virus infection promotes translocation of high mobility group box 1 protein from the nucleus to the cytosol in dendritic cells, upregulates cytokine production and modulates virus replication. J. Gen Virol. 2009, 90, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gan, C.P.; Zhang, S.; Zhou, X.K.; Li, X.F.; Wei, Y.Q.; Yang, J.L.; Wu, M. FIP200 is involved in murine pseudomonas infection by regulating HMGB1 intracellular translocation. Cell. Physiol. Biochem. 2014, 33, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.H.; Shin, J.S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 2006, 177, 7889–7897. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.W.; Jiang, W.; Reich, C.F., 3rd; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Fujii, T.; Ishijima, R.; Tachibana, H.; Yokoue, N.; Takasawa, R.; Tanuma, S.-I. The release of high mobility group box 1 in apoptosis is triggered by nucleosomal DNA fragmentation. Arch. Biochem. Biophys. 2011, 506, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; Frankel, A.E.; Thorburn, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009, 16, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Furnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; de Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: Implications for the pathogenesis of SLE. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Wang, H.; Yuan, R.; Li, H.; Ochani, M.; Ochani, K.; Rosas-Ballina, M.; Czura, C.J.; Huston, J.M.; Miller, E.; et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 2006, 203, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 2005, 174, 7506–7515. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Zuliani, P.; Komuravelli, A.; Faeder, J.R.; Clarke, E.M. Analysis and verification of the HMGB1 signaling pathway. BMC Bioinform. 2010, 11, S10. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, H.K.; Yoon, J.S.; Kim, S.J.; Kim, E.S.; Ahn, K.S.; Kim, D.S.; Yoon, S.S.; Kim, B.K.; Lee, Y.Y. Advanced glycation end product (age)-induced proliferation of hel cells via receptor for age-related signal pathways. Int. J. Oncol. 2008, 33, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Capobianco, A.; Esposito, A.; de Cobelli, F.; Canu, T.; Monno, A.; Raucci, A.; Sanvito, F.; Doglioni, C.; Nawroth, P.P.; et al. Maturing dendritic cells depend on rage for in vivo homing to lymph nodes. J. Immunol. 2008, 180, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fu, S.; Fan, X.G.; Lotze, M.T.; Zeh, H.J., 3rd; Tang, D.; Kang, R. Nuclear damp complex-mediated rage-dependent macrophage cell death. Biochem. Biophys. Res. Commun. 2015, 458, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Campana, L.; Bosurgi, L.; Rovere-Querini, P. Hmgb1: A two-headed signal regulating tumor progression and immunity. Curr. Opin. Immunol. 2008, 20, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.; Holdenrieder, S. Immunogene Zelltodmarker HMGB1 und sRAGE als neue prädiktive und prognostische Serum Biomarker bei Tumorerkrankungen/Immunogenic cell death markers HMGB1 and sRAGE as new predictive and prognostic serum biomarkers in cancer disease. LaboratoriumsMedizin 2013, 37, 29–51. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007, 220, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.H.; Oh, Y.J.; Kim, E.S.; Choi, J.E.; Shin, J.S. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J. Immunol. 2008, 180, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Shiratsuchi, A.; Watanabe, I.; Takeuchi, O.; Akira, S.; Nakanishi, Y. Inhibitory effect of toll-like receptor 4 on fusion between phagosomes and endosomes/lysosomes in macrophages. J. Immunol. 2004, 172, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. Hmgb1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 2008, 180, 2531–2537. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll like receptor 9. J. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. HMGB1 loves company. J. Leukocyte Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Tang, J.; Zheng, P.; Liu, Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 2009, 323, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Unlu, S.; Tang, S.; Wang, E.; Martinez, I.; Tang, D.; Bianchi, M.E.; Zeh, H.J., 3rd; Lotze, M.T. Damage associated molecular pattern molecule-induced microRNAs (DAMPmiRs) in human peripheral blood mononuclear cells. PLoS ONE 2012, 7, e38899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yue, Y.; Zhu, Y.; Xiong, S. Extracellular, but not intracellular HMGB1, facilitates self-DNA induced macrophage activation via promoting DNA accumulation in endosomes and contributes to the pathogenesis of lupus nephritis. Mol. Immunol. 2015, 65, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Fukami, A.; Adachi, H.; Yamagishi, S.; Matsui, T.; Ueda, S.; Nakamura, K.; Enomoto, M.; Otsuka, M.; Kumagae, S.; Nanjo, Y.; et al. Factors associated with serum high mobility group box 1 (HMGB1) levels in a general population. Metabolism 2009, 58, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Zeh Iii, H.J.; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. Biophys. Acta 2010, 1799, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet 2005, 6, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Giavara, S.; Kosmidou, E.; Hande, M.P.; Bianchi, M.E.; Morgan, A.; d’Adda di Fagagna, F.; Jackson, S.P. Yeast Nhp6A/B and mammalian Hmgb1 facilitate the maintenance of genome stability. Curr. Biol. 2005, 15, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Malina, J.; Kasparkova, J.; Natile, G.; Brabec, V. Recognition of major DNA adducts of enantiomeric cisplatin analogs by HMG box proteins and nucleotide excision repair of these adducts. Chem. Biol. 2002, 9, 629–638. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Volp, K.; Bosser, S.; Solbach, C.; Lichter, P.; Joos, S.; Zornig, M. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 2003, 17, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Billiar, T.R.; Lotze, M.T. A janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol. Med. 2012, 18, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; de Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tang, D.; Lotze, M.T. Menage a trois in stress: Damps, redox and autophagy. Semin. Cancer Biol. 2013, 23, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Chihara, Y.; Kondo, H. Differential effects between amphoterin and advanced glycation end products on colon cancer cells. Int. J. Cancer 2003, 104, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, Y.; Levy, R.M.; Fan, J.J.; Hackam, D.J.; Vodovotz, Y.; Yang, H.; Tracey, K.J.; Billiar, T.R.; Wilson, M.A. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: Role of HMGB1-TLR4 signaling. J. Immunol. 2007, 178, 6573–6580. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Lee, J.Y.; Yoon, B.K.; Bae, D.S.; Choi, D.S. Effects of HMGB-1 overexpression on cell-cycle progression in MCF-7 cells. J. Korean Med. Sci. 2004, 19, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Loux, T.; Livesey, K.M.; Billiar, T.R.; Wang, H.; Van Houten, B.; Lotze, M.T.; Zeh, H.J. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014, 33, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Wittig, R.; Coy, J.F. The role of glucose metabolism and glucose-associated signalling in cancer. Perspect. Med. Chem. 2008, 1, 64–82. [Google Scholar]
- Schlueter, C.; Weber, H.; Meyer, B.; Rogalla, P.; Roser, K.; Hauke, S.; Bullerdiek, J. Angiogenetic signaling through hypoxia: HMGB1: An angiogenetic switch molecule. Am. J. Pathol. 2005, 166, 1259–1263. [Google Scholar] [CrossRef]
- Lin, Q.; Yang, X.P.; Fang, D.; Ren, X.; Zhou, H.; Fang, J.; Liu, X.; Zhou, S.; Wen, F.; Yao, X.; et al. High-mobility group box-1 mediates Toll-like receptor 4-dependent angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of Toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Chen, Y.; Fu, X.; Zhang, L.; Tian, J.; Hao, Q. HMGB1 promotes lymphangiogenesis of human lymphatic endothelial cells in vitro. Med. Oncol. 2012, 29, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of rage-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [PubMed]
- Liu, L.; Zhao, L.; Zhang, Y.; Zhang, Q.; Ding, Y. Proteomic analysis of tiam1-mediated metastasis in colorectal cancer. Cell Biol. Int. 2007, 31, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Harada, O.; Suga, T.; Suzuki, T.; Nakamoto, K.; Kobayashi, M.; Nomiyama, T.; Nadano, D.; Ohyama, C.; Fukuda, M.N.; Nakayama, J. The role of trophinin, an adhesion molecule unique to human trophoblasts, in progression of colorectal cancer. Int. J. Cancer 2007, 121, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Aychek, T.; Miller, K.; Sagi-Assif, O.; Levy-Nissenbaum, O.; Israeli-Amit, M.; Pasmanik-Chor, M.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Witz, I.P. E-selectin regulates gene expression in metastatic colorectal carcinoma cells and enhances hmgb1 release. Int. J. Cancer 2008, 123, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ohmori, H.; Fujii, K.; Moriwaka, Y.; Sasahira, T.; Kurihara, M.; Tatsumoto, N.; Sasaki, T.; Yamashita, Y.; Kuniyasu, H. Hmgb1 attenuates anti-metastatic defence of the liver in colorectal cancer. Eur. J. Cancer 2010, 46, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Moriwaka, Y.; Luo, Y.; Ohmori, H.; Fujii, K.; Tatsumoto, N.; Sasahira, T.; Kuniyasu, H. HMGB1 attenuates anti-metastatic defense of the lymph nodes in colorectal cancer. Pathobiology 2010, 77, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through Toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Clear, A.; Liu, F.T.; Matthews, J.; Uddin, N.; McCarthy, A.; Hoxha, E.; Durance, C.; Iqbal, S.; Gribben, J.G. Extracellular hmgb1 promotes differentiation of nurse-like cells in chronic lymphocytic leukemia. Blood 2014, 123, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Messmer, D.; Yang, H.; Telusma, G.; Knoll, F.; Li, J.; Messmer, B.; Tracey, K.J.; Chiorazzi, N. High mobility group box protein 1: An endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 2004, 173, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Kusume, A.; Sasahira, T.; Luo, Y.; Isobe, M.; Nakagawa, N.; Tatsumoto, N.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Suppression of dendritic cells by HMGB1 is associated with lymph node metastasis of human colon cancer. Pathobiology 2009, 76, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Falo, L.D., Jr.; You, Z. Knockdown of hmgb1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J. Immunol. 2011, 187, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.Y.; Wang, D.Y.; Tanaka, M.; Suzuki, M.; Kiyokawa, E.; Igarashi, H.; Naito, Y.; Shen, Q.; Sugimura, H. Expression of high-mobility group-1 mrna in human gastrointestinal adenocarcinoma and corresponding non-cancerous mucosa. Int. J. Cancer 1997, 74, 1–6. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; van Houten, B.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011, 13, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Abdulahad, D.A.; Westra, J.; Bijzet, J.; Limburg, P.C.; Kallenberg, C.G.; Bijl, M. High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, R71. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Jiao, Y.; Cui, B.; Gao, X.; Xia, Y.; Zhao, Y. Immune complexes activate human endothelium involving the cell-signaling HMGB1-RAGE axis in the pathogenesis of lupus vasculitis. Lab. Invest. 2013, 93, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Kawahara, K.; Yone, K.; Hashiguchi, T.; Yamakuchi, M.; Goto, M.; Inoue, K.; Yamada, S.; Ijiri, K.; Matsunaga, S.; et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003, 48, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, S.W.; Kim, H.Y.; Lee, W.S.; Hong, K.W.; Kim, C.D. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1α activation. Eur. J. Immunol. 2015, 45, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Yamoah, K.; Brebene, A.; Baliram, R.; Inagaki, K.; Dolios, G.; Arabi, A.; Majeed, R.; Amano, H.; Wang, R.; Yanagisawa, R.; et al. High-mobility group box proteins modulate tumor necrosis factor-alpha expression in osteoclastogenesis via a novel deoxyribonucleic acid sequence. Mol. Endocrinol. 2008, 22, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Han, J.Y.; Xi, C.X.; Xie, J.X.; Feng, X.; Wang, C.Y.; Mei, L.; Xiong, W.C. HMGB1 regulates RANKL-induced osteoclastogenesis in a manner dependent on RAGE. J. Bone Miner Res. 2008, 23, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundback, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Yan, W.; Geczy, C.L.; Brown, M.A.; Thomas, R. Serum levels of soluble receptor for advanced glycation end products and of s100 proteins are associated with inflammatory, autoantibody, and classical risk markers of joint and vascular damage in rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R39. [Google Scholar] [CrossRef] [PubMed]
- Chuong, C.; Katz, J.; Pauley, K.M.; Bulosan, M.; Cha, S. Rage expression and NF-κB activation attenuated by extracellular domain of rage in human salivary gland cell line. J. Cell. Physiol. 2009, 221, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Nadeau, B.A.; Day, D.E.; Huber-Lang, M.; Mackay, C.R.; Zetoune, F.S.; Gerard, N.P.; Cianflone, K.; Kohl, J.; et al. Functional roles for C5a receptors in sepsis. Nat. Med. 2008, 14, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Burt, B.M.; Nguyen, H.M.; Bamboat, Z.M.; DeMatteo, R.P. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J. Exp. Med. 2008, 205, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.H.; Nho, D.H.; Song, R.H.; Kim, S.H.; Lee, M.J.; Nemzek, J.A.; Park, J. High-mobility group box 1 as a surrogate prognostic marker in dogs with systemic inflammatory response syndrome. J. Vet. Emerg. Crit. Care 2010, 20, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Kornblit, B.; Munthe-Fog, L.; Madsen, H.O.; Strom, J.; Vindelov, L.; Garred, P. Association of hmgb1 polymorphisms with outcome in patients with systemic inflammatory response syndrome. Crit. Care 2008, 12, R83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zedler, S.; Faist, E. The impact of endogenous triggers on trauma-associated inflammation. Curr. Opin. Crit. Care 2006, 12, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit. Care 2009, 13, R174. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Karki, A.; Du, D.Y.; Zhao, X.J.; Xiang, X.Y.; Lu, Z.Q. Plasma levels of high mobility group box 1 increase in patients with posttraumatic stress disorder after severe blunt chest trauma: A prospective cohort study. J. Surg. Res. 2015, 193, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Lantos, J.; Foldi, V.; Roth, E.; Weber, G.; Bogar, L.; Csontos, C. Burn trauma induces early HMGB1 release in patients: Its correlation with cytokines. Shock 2010, 33, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, V.; Signore, M.; Pacini, I.; Misasi, R.; Tellan, G.; Garofalo, T.; Lococo, E.; Chirletti, P.; Sorice, M.; Delogu, G. Increased HMGB1 expression and release by mononuclear cells following surgical/anesthesia trauma. Crit. Care 2010, 14, R197. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Unraveling the role of high mobility group box protein 1 in severe trauma. Crit. Care 2009, 13, 1004. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Lim, B.J.; Park, S.; Choi, D.; Kim, H.W.; Ku, N.S.; Han, S.H.; Kim, C.O.; Choi, J.Y.; Song, Y.G.; et al. The effect of sRAGE-Fc fusion protein attenuates inflammation and decreases mortality in a murine cecal ligation and puncture model. Inflamm. Res. 2012, 61, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Furnrohr, B.G.; Weber, C.; Haslbeck, M.; Wilhelm, S.; Herrmann, M.; Voll, R.E. Factors masking HMGB1 in human serum and plasma. J. Leukoc Biol. 2007, 81, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Barnay-Verdier, S.; Gaillard, C.; Messmer, M.; Borde, C.; Gibot, S.; Marechal, V. PCA-ELISA: A sensitive method to quantify free and masked forms of HMGB1. Cytokine 2011, 55, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, C.; Borde, C.; Gozlan, J.; Marechal, V.; Strauss, F. A high-sensitivity method for detection and measurement of HMGB1 protein concentration by high-affinity binding to DNA hemicatenanes. PLoS ONE 2008, 3, e2855. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.; Lehner, J.; Fersching, D.; Siegele, B.; Stoetzer, O.J.; Holdenrieder, S. Methodological and preanalytical evaluation of a RAGE immunoassay. Anticancer Res. 2012, 32, 2075–2078. [Google Scholar] [PubMed]
- Kim, J.; Sohn, E.; Kim, C.S.; Jo, K.; Kim, J.S. The role of high-mobility group box-1 protein in the development of diabetic nephropathy. Am. J. Nephrol. 2011, 33, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Humpert, P.M.; Papadopoulos, G.; Schaefer, K.; Djuric, Z.; Konrade, I.; Morcos, M.; Nawroth, P.P.; Bierhaus, A. Srage and esrage are not associated with peripheral or autonomic neuropathy in type 2 diabetes. Horm. Metab. Res. 2007, 39, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N.; Matsuhisa, M.; Kaneto, H.; Matsuoka, T.A.; Sakamoto, K.; Yasuda, T.; Yamasaki, Y. Endogenous secretory rage but not soluble rage is associated with carotid atherosclerosis in type 1 diabetes patients. Diabetes Vasc. Dis. Res. 2008, 5, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.Y.; Jiao, Y.L.; Zhang, J.; Yang, Q.R.; Zhang, Z.F.; Shen, Y.J.; Chen, Z.J.; Zhao, Y.R. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-α and TNF-α in systemic lupus erythematosus. Rheumatol. Int. 2012, 32, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.Y.; Ma, J.L.; Jiao, Y.L.; Li, J.F.; Wang, L.C.; Yang, Q.R.; You, L.; Cui, B.; Chen, Z.J.; Zhao, Y.R. The plasma level of soluble receptor for advanced glycation end products is decreased in patients with systemic lupus erythematosus. Scand. J. Immunol. 2012, 75, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Sundberg, E.; Ulfgren, A.K.; Palmblad, K.; Li, J.; Wang, H.; Ulloa, L.; Yang, H.; Yan, X.J.; Furie, R.; et al. High mobility group box chromosomal protein 1: A novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002, 46, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Li, J.; Sundberg, E.; Aveberger, A.C.; Palmblad, K.; Yang, H.; Tracey, K.J.; Andersson, U.; Harris, H.E. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003, 48, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.S.; Bruchfeld, A.; Yang, L.; Qureshi, A.R.; Gallowitsch-Puerta, M.; Patel, N.B.; Huston, B.J.; Chavan, S.; Rosas-Ballina, M.; Gregersen, P.K.; et al. Cholinergic anti-inflammatory pathway activity and high mobility group box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol. Med. 2007, 13, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.S.; Shang, Q.; Li, E.K.; Wong, S.; Li, R.J.; Lee, K.L.; Leung, Y.Y.; Ying, K.Y.; Yim, C.W.; Kun, E.W.; et al. Serum soluble receptor for advanced glycation end products levels and aortic augmentation index in early rheumatoid arthritis—A prospective study. Semin. Arthritis Rheum. 2013, 42, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Myles, A.; Viswanath, V.; Singh, Y.P.; Aggarwal, A. Soluble receptor for advanced glycation endproducts is decreased in patients with juvenile idiopathic arthritis (ERA category) and inversely correlates with disease activity and S100A12 levels. J. Rheumatol. 2011, 38, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Bruchfeld, A.; Wendt, M.; Bratt, J.; Qureshi, A.R.; Chavan, S.; Tracey, K.J.; Palmblad, K.; Gunnarsson, I. High-mobility group box-1 protein (HMGB1) is increased in antineutrophilic cytoplasmatic antibody (ANCA)-associated vasculitis with renal manifestations. Mol. Med. 2011, 17, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.K.; Cha, H.S.; Bae, E.K.; Lee, J.; Koh, E.M. Extracellular high-mobility group box 1 is increased in patients with behcet’s disease with intestinal involvement. J. Korean Med. Sci. 2011, 26, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Wittkowski, H.; Hirono, K.; Ichida, F.; Vogl, T.; Ye, F.; Yanlin, X.; Saito, K.; Uese, K.; Miyawaki, T.; Viemann, D.; et al. Acute Kawasaki disease is associated with reverse regulation of soluble receptor for advance glycation end products and its proinflammatory ligand S100A12. Arthritis Rheum. 2007, 56, 4174–4181. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gong, F.; Zhang, Q.; Xie, C.; Wang, W.; Fu, S. Reverse regulation of soluble receptor for advanced glycation end products and proinflammatory factor resistin and S100A12 in Kawasaki disease. Arthritis Res. Ther. 2012, 14, R251. [Google Scholar] [CrossRef] [PubMed]
- Takahata, R.; Ono, S.; Tsujimoto, H.; Hiraki, S.; Kimura, A.; Kinoshita, M.; Miyazaki, H.; Saitoh, D.; Hase, K. Postoperative serum concentrations of high mobility group box chromosomal protein-1 correlates to the duration of sirs and pulmonary dysfunction following gastrointestinal surgery. J. Surg. Res. 2011, 170, e135–e140. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Carles, M.; Brohi, K.; Calfee, C.S.; Rahn, P.; Call, M.S.; Chesebro, B.B.; West, M.A.; Pittet, J.F. Early release of soluble receptor for advanced glycation endproducts after severe trauma in humans. J. Trauma 2010, 68, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Asgeirsson, T.; Zhang, S.; Khoo, S.K.; Resau, J.H.; Dujovny, N.; Senagore, A.J. Serum adiponectin, resistin, and circulating soluble receptor for advanced glycation end products in colectomy patients. Mediat. Inflamm. 2011, 2011, 916807. [Google Scholar] [CrossRef] [PubMed]
- Punyadeera, C.; Schneider, E.M.; Schaffer, D.; Hsu, H.Y.; Joos, T.O.; Kriebel, F.; Weiss, M.; Verhaegh, W.F. A biomarker panel to discriminate between systemic inflammatory response syndrome and sepsis and sepsis severity. J. Emerg. Trauma Shock 2010, 3, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.J.; Bellamy, S.L.; Localio, A.R.; Wickersham, N.; Diamond, J.M.; Weinacker, A.; Lama, V.N.; Bhorade, S.; Belperio, J.A.; Crespo, M.; et al. A panel of lung injury biomarkers enhances the definition of primary graft dysfunction (PGD) after lung transplantation. J. Heart Lung Transplant. 2012, 31, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Lee, S.-G.; Kim, H.; Hong, D.; Chung, J.; Stroncek, D.; Lim, J.-B. Serum high mobility group box-1 (HMGB1) is closely associated with the clinical and pathologic features of gastric cancer. J. Translat. Med. 2009, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.Q.; Jia, C.Q.; Liu, C.T.; Lu, X.F.; Zhong, N.; Zhang, Z.L.; Fan, W.; Li, Y.Q. Serum high mobility group box chromosomal protein 1 is associated with clinicopathologic features in patients with hepatocellular carcinoma. Dig. Liver Dis. 2008, 40, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.H.; Jia, C.Q.; Tian, H.; Xiao, W.; Li, Y.; Wang, A.H.; Dong, L.; Lin, D.J. Serum high mobility group box protein 1 as a clinical marker for non-small cell lung cancer. Respir. Med. 2009, 103, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Du, X.; Zhang, X.; Li, D.; Lu, C.; Li, Q.; Ma, Z.; Song, Q.; Wang, C. Clinical value of serum HMGB1 levels in early detection of recurrent squamous cell carcinoma of uterine cervix: Comparison with serum SCCA, CYFRA21–1, and CEA levels. Croat. Med. J. 2009, 50, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.; Boeck, S.; Heinemann, V.; Haas, M.; Stieber, P.; Nagel, D.; Holdenrieder, S. Circulating nucleosomes and immunogenic cell death markers HMGB1, sRAGE and DNAse in patients with advanced pancreatic cancer undergoing chemotherapy. Int. J. Cancer 2013, 133, 2619–2630. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.L.; Cao, L.Z.; Yu, Y.; Zhang, G.Y.; Xiao, X.Z. High mobility group box 1 is increased in children with acute lymphocytic leukemia and stimulates the release of tumor necrosis factor-alpha in leukemic cell. Zhonghua Er Ke Za Zhi 2007, 45, 329–333. [Google Scholar] [PubMed]
- Candolfi, M.; Yagiz, K.; Foulad, D.; Alzadeh, G.E.; Tesarfreund, M.; Muhammad, A.K.M.G.; Puntel, M.; Kroeger, K.M.; Liu, C.; Lee, S.; et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: Efficacy and neurotoxicity. Clin. Cancer Res. 2009, 15, 4401–4414. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; DeMarco, R.A.; Mailliard, R.B.; Han, J.; Rabinowich, H.; Kalinski, P.; Stolz, D.B.; Zeh, H.J., 3rd; Lotze, M.T. Cytolytic cells induce HMGB1 release from melanoma cell lines. J. Leukoc Biol. 2007, 81, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Dong Xda, E.; Ito, N.; Lotze, M.T.; Demarco, R.A.; Popovic, P.; Shand, S.H.; Watkins, S.; Winikoff, S.; Brown, C.K.; Bartlett, D.L.; et al. High mobility group box I (HMGB1) release from tumor cells after treatment: Implications for development of targeted chemoimmunotherapy. J. Immunother. 2007, 30, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Kohles, N.; Nagel, D.; Jungst, D.; Stieber, P.; Holdenrieder, S. Predictive value of immunogenic cell death biomarkers HMGB1, sRAGE, and DNAse in liver cancer patients receiving transarterial chemoembolization therapy. Tumour. Biol. 2012, 33, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Fahmueller, Y.N.; Nagel, D.; Hoffmann, R.T.; Tatsch, K.; Jakobs, T.; Stieber, P.; Holdenrieder, S. Immunogenic cell death biomarkers HMGB1, RAGE, and DNAse indicate response to radioembolization therapy and prognosis in colorectal cancer patients. Int. J. Cancer 2013, 132, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Stoetzer, O.J.; Fersching, D.M.; Salat, C.; Steinkohl, O.; Gabka, C.J.; Hamann, U.; Braun, M.; Feller, A.M.; Heinemann, V.; Siegele, B.; et al. Prediction of response to neoadjuvant chemotherapy in breast cancer patients by circulating apoptotic biomarkers nucleosomes, DNAse, cytokeratin-18 fragments and survivin. Cancer Lett. 2013, 336, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Stoetzer, O.J.; Fersching, D.M.; Salat, C.; Steinkohl, O.; Gabka, C.J.; Hamann, U.; Braun, M.; Feller, A.M.; Heinemann, V.; Siegele, B.; et al. Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumour Biol. 2013, 34, 81–90. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilzweger, C.; Holdenrieder, S. Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases. Diagnostics 2015, 5, 219-253. https://doi.org/10.3390/diagnostics5020219
Pilzweger C, Holdenrieder S. Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases. Diagnostics. 2015; 5(2):219-253. https://doi.org/10.3390/diagnostics5020219
Chicago/Turabian StylePilzweger, Christin, and Stefan Holdenrieder. 2015. "Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases" Diagnostics 5, no. 2: 219-253. https://doi.org/10.3390/diagnostics5020219
APA StylePilzweger, C., & Holdenrieder, S. (2015). Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases. Diagnostics, 5(2), 219-253. https://doi.org/10.3390/diagnostics5020219