Prenatal Diagnosis of Autosomal Recessive Renal Tubular Dysgenesis with Anhydramnios Caused by a Mutation in the AGT Gene

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
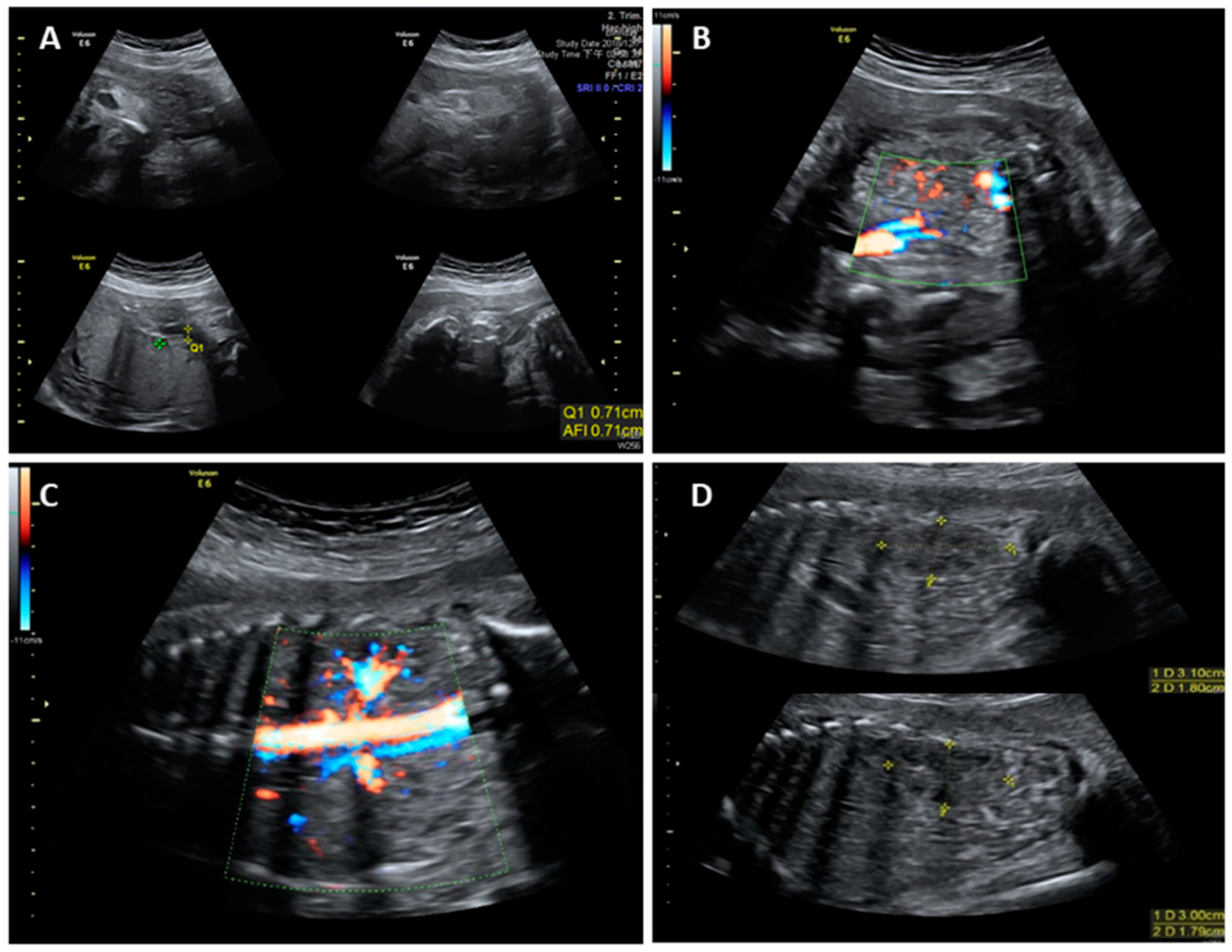
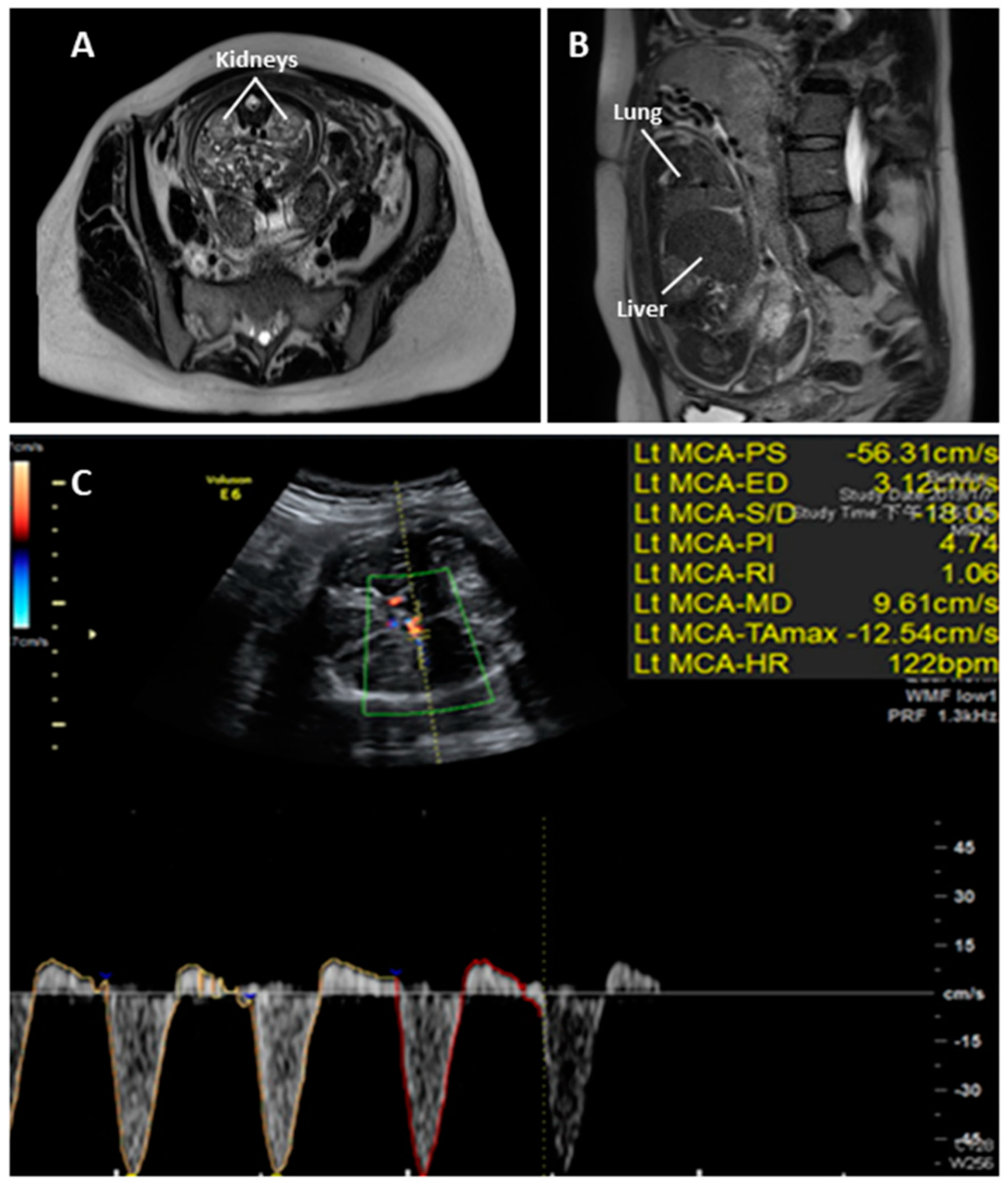
2. Case Report
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allanson, J.E.; Pantzar, J.T.; MacLeod, P.M. Possible new autosomal recessive syndrome with unusual renal histopathological changes. Am. J. Med. Genet. 1983, 16, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Gubler, M.C. Renal tubular dysgenesis. Pediatr. Nephrol. 2014, 29, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Moss, G.; Primhak, R.; Coombs, R. Congenital renal tubular dysplasia and skull ossification defects similar to teratogenic effects of angiotensin converting enzyme (ACE) inhibitors. J. Med. Genet. 1997, 34, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Kriegsmann, J.; Coerdt, W.; Kommoss, F.; Beetz, R.; Hallermann, C.; Müntefering, H. Renal tubular dysgenesis (RTD). An important cause of the oligohydramnion-sequence. Report of 3 cases and review of the literature. Pathol. Res. Pract. 2000, 196, 861–865. [Google Scholar] [CrossRef]
- Lacoste, M.; Cai, Y.; Guicharnaud, L.; Mounier, F.; Dumez, Y.; Bouvier, R.; Dijoud, F.; Gonzales, M.; Chatten, J.; Delezoide, A.L.; et al. Renal tubular dysgenesis, a not uncommon autosomal recessive disorder leading to oligohydramnios: Role of the Renin-Angiotensin system. J. Am. Soc. Nephrol. 2006, 17, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F. Potter’s syndrome: A study of 15 patients. Arch. Iranian. Med. 2004, 7, 186–189. [Google Scholar]
- Shastry, S.M.; Kolte, S.S.; Sanagapati, P.R. Potter’s Sequence. J. Clin. Neonatol. 2012, 1, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Barr, M., Jr.; Sedman, A.B.; Heidelberger, K.P. Renal tubular dysgenesis in twins. Pedi. Atr. Nephrol. 1998, 12, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Mahieu-Caputo, D.; Dommergues, M.; Delezoide, A.L.; Lacoste, M.; Cai, Y.; Narcy, F.; Jolly, D.; Gonzales, M.; Dumez, Y.; Gubler, M.C. Twin-to-twin transfusion syndrome. Role of the fetal renin-angiotensin system. Am. J. Pathol. 2000, 156, 629–636. [Google Scholar] [CrossRef]
- Barr, M., Jr. Teratogen update: Angiotensin-converting enzyme inhibitors. Teratology 1994, 50, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Martinovic, J.; Benachi, A.; Laurent, N.; Daikha-Dahmane, F.; Gubler, M.C. Fetal toxic effects and angiotensin-II-receptor antagonists. Lancet 2001, 358, 241–242. [Google Scholar] [CrossRef]
- Saji, H.; Yamanaka, M.; Hagiwara, A.; Ijiri, R. Losartan and fetal toxic effects. Lancet 2001, 357, 363. [Google Scholar] [CrossRef]
- Gribouval, O.; Gonzales, M.; Neuhaus, T.; Aziza, J.; Bieth, E.; Laurent, N.; Bouton, J.M.; Feuillet, F.; Makni, S.; Ben Amar, H.; et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat. Genet. 2005, 37, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Gribouval, O.; Morinière, V.; Pawtowski, A.; Arrondel, C.; Sallinen, S.L.; Saloranta, C.; Clericuzio, C.; Viot, G.; Tantau, J.; Blesson, S.; et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat. 2012, 33, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. N. Am. J. Med. Sci. (Boston) 2011, 4, 183–190, PMCID:PMC3291105. [Google Scholar] [CrossRef] [PubMed]
- Barr, M., Jr.; Cohen, M.M., Jr. ACE inhibitor fetopathy and hypocalvaria: The kidney-skull connection. Teratology 1991, 44, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Van Vuuren, S.H.; Damen-Elias, H.A.; Stigter, R.H.; van der Doef, R.; Goldschmeding, R.; de Jong, T.P.; Westers, P.; Visser, G.H.; Pistorius, L.R. Size and volume charts of fetal kidney, renal pelvis and adrenal gland. Ultrasound Obstet. Gynecol. 2012, 40, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Moldavsky, M. Renal tubular dysgenesis in Israel: Pathologist’s experience and literature review. Isr. Med. Assoc. J. 2009, 11, 6–10. [Google Scholar] [PubMed]
- Ramalho, C.; Matias, A.; Brandão, O.; Montenegro, N. Renal tubular dysgenesis: Report of two cases in a non-consanguineous couple and review of the literature. Fetal Diagn. Ther. 2007, 22, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Cavoretto, P. Prediction of pulmonary hypoplasia in mid-trimester preterm prelabor rupture of membranes: Research or clinical practice? Ultrasound Obstet. Gynecol. 2012, 39, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Van Teeffelen, A.S.; Van Der Heijden, J.; Oei, S.G.; Porath, M.M.; Willekes, C.; Opmeer, B.; Mol, B.W. Accuracy of imaging parameters in the prediction of lethal pulmonary hypoplasia secondary to mid-trimester prelabor rupture of fetal membranes: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2012, 39, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Brownfoot, F.C.; Cluver, C.A.; Walker, S.P. Persistent reversed end diastolic flow in the fetal middle cerebral artery: An ominous finding. Ultrasound 2015, 23, 186–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, G.-C.; Chen, Y.-C.; Wu, W.-J.; Chang, S.-P.; Chang, T.-Y.; Lin, W.-H.; Chen, M. Prenatal Diagnosis of Autosomal Recessive Renal Tubular Dysgenesis with Anhydramnios Caused by a Mutation in the AGT Gene. Diagnostics 2019, 9, 185. https://doi.org/10.3390/diagnostics9040185
Ma G-C, Chen Y-C, Wu W-J, Chang S-P, Chang T-Y, Lin W-H, Chen M. Prenatal Diagnosis of Autosomal Recessive Renal Tubular Dysgenesis with Anhydramnios Caused by a Mutation in the AGT Gene. Diagnostics. 2019; 9(4):185. https://doi.org/10.3390/diagnostics9040185
Chicago/Turabian StyleMa, Gwo-Chin, Ying-Chung Chen, Wan-Ju Wu, Shun-Ping Chang, Ting-Yu Chang, Wen-Hsiang Lin, and Ming Chen. 2019. "Prenatal Diagnosis of Autosomal Recessive Renal Tubular Dysgenesis with Anhydramnios Caused by a Mutation in the AGT Gene" Diagnostics 9, no. 4: 185. https://doi.org/10.3390/diagnostics9040185
APA StyleMa, G.-C., Chen, Y.-C., Wu, W.-J., Chang, S.-P., Chang, T.-Y., Lin, W.-H., & Chen, M. (2019). Prenatal Diagnosis of Autosomal Recessive Renal Tubular Dysgenesis with Anhydramnios Caused by a Mutation in the AGT Gene. Diagnostics, 9(4), 185. https://doi.org/10.3390/diagnostics9040185