Evaluation of Phenotypic and Genotypic Variations of Drug Metabolising Enzymes and Transporters in Chronic Pain Patients Facing Adverse Drug Reactions or Non-Response to Analgesics: A Retrospective Study

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients and Setting
2.2. Evaluation Criteria
2.3. Explored Pathways and Metabolic Status
2.3.1. Genotyping
2.3.2. Phenotyping
2.3.3. Statistical Analysis
3. Results
3.1. Patients, ADR or Non-Response, and Involved Analgesic Drugs
3.2. Metabolic Status
3.3. Concordance Between Predicted and Measured CYP Activity
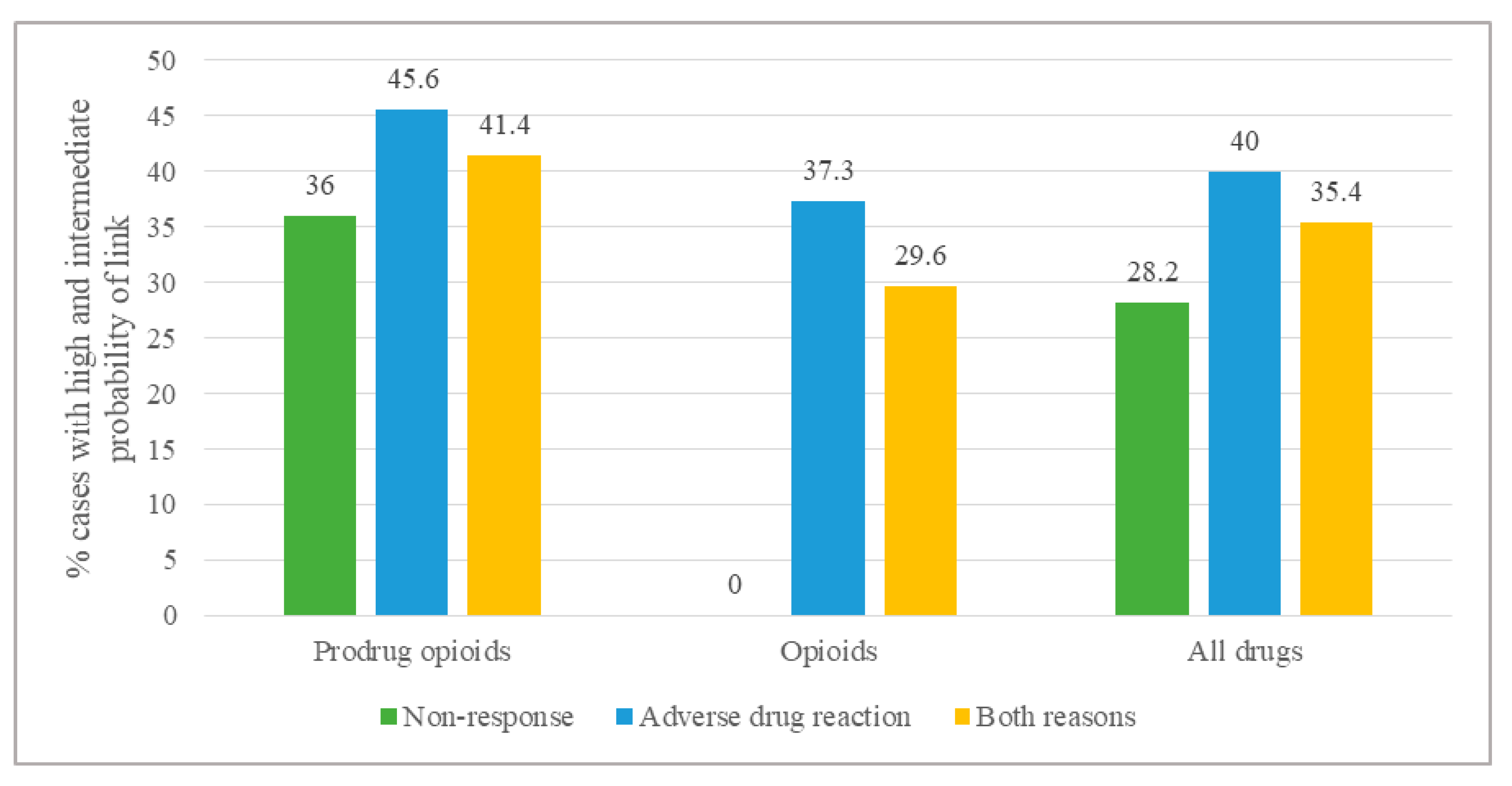
3.4. Link Between Metabolic Status and ADR or Non-Response
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Type of Demand | Probability of Link According to Phenotype for Major (Minor) Metabolic Pathway | |||
|---|---|---|---|---|
| UM | NM | IM | PM | |
| Non-response to parent compound | 2 (1) | 0 | 0 | 0 |
| Non-response to active metabolite | 0 | 0 | 1 (0) | 2 (1) |
| Adverse reaction to parent compound | 0 | 0 | 1 (0) | 2 (1) |
| Adverse reaction to active metabolite | 2 (1) | 0 | 0 | 0 |
References
- Jara, C.; Del Barco, S.; Grávalos, C.; Hoyos, S.; Hernández, B.; Muñoz, M.; Quintanar, T.; Meana, J.A.; Rodriguez, C.; de Las Peñas, R. SEOM clinical guideline for treatment of cancer pain (2017). Clin. Transl. Oncol. 2018, 20, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attal, N.; Cruccu, G.; Baron, R.; Haanpää, M.; Hansson, P.; Jensen, T.S.; Nurmikko, T. European Federation of Neurological Societies EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur. J. Neurol. 2010, 17, 1113-e88. [Google Scholar] [CrossRef] [PubMed]
- Cruccu, G.; Gronseth, G.; Alksne, J.; Argoff, C.; Brainin, M.; Burchiel, K.; Nurmikko, T.; Zakrzewska, J.M. American Academy of Neurology Society; European Federation of Neurological Society AAN-EFNS guidelines on trigeminal neuralgia management. Eur. J. Neurol. 2008, 15, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Kaye, A.D.; Urman, R.D. Pharmacogenomics in Pain Management. Anesthesiol. Clin. 2017, 35, 295–304. [Google Scholar] [CrossRef]
- Senagore, A.J.; Champagne, B.J.; Dosokey, E.; Brady, J.; Steele, S.R.; Reynolds, H.L.; Stein, S.L.; Delaney, C.P. Pharmacogenetics-guided analgesics in major abdominal surgery: Further benefits within an enhanced recovery protocol. Am. J. Surg. 2017, 213, 467–472. [Google Scholar] [CrossRef]
- Rollason, V.; Samer, C.F.; Daali, Y.; Desmeules, J.A. Prediction by pharmacogenetics of safety and efficacy of non-steroidal anti- inflammatory drugs: A review. Curr. Drug Metab. 2014, 15, 326–343. [Google Scholar] [CrossRef]
- Phillips, C.J. The Cost and Burden of Chronic Pain. Rev. Pain 2009, 3, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Maniadakis, N.; Gray, A. The economic burden of back pain in the UK. Pain 2000, 84, 95–103. [Google Scholar] [CrossRef]
- Stewart, W.F.; Ricci, J.A.; Chee, E.; Morganstein, D.; Lipton, R. Lost productive time and cost due to common pain conditions in the US workforce. JAMA 2003, 290, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Lloret-Linares, C.; Rollason, V.; Lorenzini, K.I.; Samer, C.; Daali, Y.; Gex-Fabry, M.; Aubry, J.-M.; Desmeules, J.; Besson, M. Screening for genotypic and phenotypic variations in CYP450 activity in patients with therapeutic problems in a psychiatric setting, a retrospective study. Pharmacol. Res. 2017, 118, 104–110. [Google Scholar] [CrossRef]
- Samer, C.F.; Lorenzini, K.I.; Rollason, V.; Daali, Y.; Desmeules, J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 2013, 17, 165–184. [Google Scholar] [CrossRef] [Green Version]
- DrugBank. Available online: https://go.drugbank.com/ (accessed on 12 October 2018).
- PharmVar. Available online: https://www.pharmvar.org/ (accessed on 12 October 2018).
- Hooten, W.M.; Biernacka, J.M.; O’Brien, T.G.; Cunningham, J.M.; Black, J.L. Associations of catechol-O-methyltransferase (rs4680) single nucleotide polymorphisms with opioid use and dose among adults with chronic pain. Pain 2019, 160, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Schmider, J.; Greenblatt, D.J.; von Moltke, L.L.; Harmatz, J.S.; Shader, R.I. N-demethylation of amitriptyline in vitro: Role of cytochrome P-450 3A (CYP3A) isoforms and effect of metabolic inhibitors. J. Pharmacol. Exp. Ther. 1995, 275, 592–597. [Google Scholar] [PubMed]
- Ghahramani, P.; Ellis, S.W.; Lennard, M.S.; Ramsay, L.E.; Tucker, G.T. Cytochromes P450 mediating the N-demethylation of amitriptyline. Br. J. Clin. Pharmacol. 1997, 43, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatakrishnan, K.; Greenblatt, D.J.; von Moltke, L.L.; Schmider, J.; Harmatz, J.S.; Shader, R.I. Five distinct human cytochromes mediate amitriptyline N-demethylation in vitro: Dominance of CYP 2C19 and 3A4. J. Clin. Pharmacol. 1998, 38, 112–121. [Google Scholar] [CrossRef]
- Venkatakrishnan, K.; Schmider, J.; Harmatz, J.S.; Ehrenberg, B.L.; von Moltke, L.L.; Graf, J.A.; Mertzanis, P.; Corbett, K.E.; Rodriguez, M.C.; Shader, R.I.; et al. Relative contribution of CYP3A to amitriptyline clearance in humans: In vitro and in vivo studies. J. Clin. Pharmacol. 2001, 41, 1043–1054. [Google Scholar] [CrossRef]
- Abaut, A.Y.; Chevanne, F.; Le Corre, P. Influence of efflux transporters on liver, bile and brain disposition of amitriptyline in mice. Int. J. Pharm. 2009, 378, 80–85. [Google Scholar] [CrossRef]
- Abaut, A.-Y.; Chevanne, F.; Le Corre, P. Oral bioavailability and intestinal secretion of amitriptyline: Role of P-glycoprotein? Int. J. Pharm. 2007, 330, 121–128. [Google Scholar] [CrossRef]
- Uhr, M.; Steckler, T.; Yassouridis, A.; Holsboer, F. Penetration of amitriptyline, but not of fluoxetine, into brain is enhanced in mice with blood-brain barrier deficiency due to mdr1a P-glycoprotein gene disruption. Neuropsychopharmacology 2000, 22, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.; Muriel, C.; Labrador, J.; Gonzalez-Porras, J.R.; Gonzalez-Sarmiento, R.; Lozano, F.S. Influence of UGT2B7, CYP3A4, and OPRM1 Gene Polymorphisms on Transdermal Buprenorphine Pain Control in Patients with Critical Lower Limb Ischemia Awaiting Revascularization. Pain Pract. 2016, 16, 842–849. [Google Scholar] [CrossRef]
- Brown, S.M.; Campbell, S.D.; Crafford, A.; Regina, K.J.; Holtzman, M.J.; Kharasch, E.D. P-glycoprotein is a major determinant of norbuprenorphine brain exposure and antinociception. J. Pharmacol. Exp. Ther. 2012, 343, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, N.M.; McLachlan, A.J.; Day, R.O.; Williams, K.M. Clinical pharmacokinetics and pharmacodynamics of celecoxib: A selective cyclo-oxygenase-2 inhibitor. Clin. Pharm. 2000, 38, 225–242. [Google Scholar] [CrossRef]
- Paulson, S.K.; Hribar, J.D.; Liu, N.W.; Hajdu, E.; Bible, R.H.; Piergies, A.; Karim, A. Metabolism and excretion of [(14)C] celecoxib in healthy male volunteers. Drug Metab. Dispos. 2000, 28, 308–314. [Google Scholar] [PubMed]
- Nielsen, K.K.; Brøsen, K.; Hansen, M.G.; Gram, L.F. Single-dose kinetics of clomipramine: Relationship to the sparteine and S-mephenytoin oxidation polymorphisms. Clin. Pharmacol. Ther. 1994, 55, 518–527. [Google Scholar] [CrossRef]
- Tacke, U.; Leinonen, E.; Lillsunde, P.; Seppälä, T.; Arvela, P.; Pelkonen, O.; Ylitalo, P. Debrisoquine hydroxylation phenotypes of patients with high versus low to normal serum antidepressant concentrations. J. Clin. Psychopharmacol. 1992, 12, 262–267. [Google Scholar] [PubMed]
- Nielsen, K.K.; Flinois, J.P.; Beaune, P.; Brøsen, K. The biotransformation of clomipramine in vitro, identification of the cytochrome P450s responsible for the separate metabolic pathways. J. Pharmacol. Exp. Ther. 1996, 277, 1659–1664. [Google Scholar]
- Dayer, P.; Desmeules, J.; Leemann, T.; Striberni, R. Bioactivation of the narcotic drug codeine in human liver is mediated by the polymorphic monooxygenase catalyzing debrisoquine 4-hydroxylation (cytochrome P-450 dbl/bufI). Biochem. Biophys. Res. Commun. 1988, 152, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.; Woodall, K.L.; Solbeck, P.; Ross, C.J.D.; Carleton, B.C.; Hayden, M.R.; Koren, G.; Madadi, P. Codeine-related deaths: The role of pharmacogenetics and drug interactions. Forensic Sci. Int. 2014, 239, 50–56. [Google Scholar] [CrossRef]
- Caraco, Y.; Tateishi, T.; Guengerich, F.P.; Wood, A.J. Microsomal codeine N-demethylation: Cosegregation with cytochrome P4503A4 activity. Drug Metab. Dispos. 1996, 24, 761–764. [Google Scholar]
- Yu, A.; Haining, R.L. Comparative contribution to dextromethorphan metabolism by cytochrome P450 isoforms in vitro: Can dextromethorphan be used as a dual probe for both CTP2D6 and CYP3A activities? Drug Metab. Dispos. 2001, 29, 1514–1520. [Google Scholar]
- Gorski, J.C.; Jones, D.R.; Wrighton, S.A.; Hall, S.D. Characterization of dextromethorphan N-demethylation by human liver microsomes. Contribution of the cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994, 48, 173–182. [Google Scholar] [CrossRef]
- Tang, W.; Stearns, R.A.; Wang, R.W.; Chiu, S.H.; Baillie, T.A. Roles of human hepatic cytochrome P450s 2C9 and 3A4 in the metabolic activation of diclofenac. Chem. Res. Toxicol. 1999, 12, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Bort, R.; Macé, K.; Boobis, A.; Gómez-Lechón, M.J.; Pfeifer, A.; Castell, J. Hepatic metabolism of diclofenac: Role of human CYP in the minor oxidative pathways. Biochem. Pharmacol. 1999, 58, 787–796. [Google Scholar] [CrossRef]
- Theken, K.N.; Lee, C.R.; Gong, L.; Caudle, K.E.; Formea, C.M.; Gaedigk, A.; Klein, T.E.; Agúndez, J.A.G.; Grosser, T. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2C9 and Nonsteroidal Anti-Inflammatory Drugs. Clin. Pharmacol. Ther. 2020, 108, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Bergstrom, R.F.; Reddy, S.; Quinlan, T.; Chappell, J.; Hong, Q.; Ring, B.; Knadler, M.P. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharm. 2008, 47, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.H.; Kuan, H.-Y.; Pan, A.; Sathirakul, K.; Knadler, M.P.; Gonzales, C.R.; Yeo, K.P.; Reddy, S.; Lim, M.; Ayan-Oshodi, M.; et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin. Pharmacol. Ther. 2003, 73, 170–177. [Google Scholar] [CrossRef]
- Feierman, D.E.; Lasker, J.M. Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4. Drug Metab. Dispos. 1996, 24, 932–939. [Google Scholar]
- Wandel, C.; Kim, R.; Wood, M.; Wood, A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology 2002, 96, 913–920. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Li, W.; Traeger, S.C.; Wang, B.; Cui, D.; Zhang, H.; Wen, B.; Rodrigues, A.D. Confirmation that cytochrome P450 2C8 (CYP2C8) plays a minor role in (S)-(+)- and (R)-(-)-ibuprofen hydroxylation in vitro. Drug Metab. Dispos. 2008, 36, 2513–2522. [Google Scholar] [CrossRef] [Green Version]
- Mazaleuskaya, L.L.; Theken, K.N.; Gong, L.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Ibuprofen pathways. Pharm. Genom. 2015, 25, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Koyama, E.; Sohn, D.R.; Shin, S.G.; Chiba, K.; Shin, J.G.; Kim, Y.H.; Echizen, H.; Ishizaki, T. Metabolic disposition of imipramine in oriental subjects: Relation to metoprolol alpha-hydroxylation and S-mephenytoin 4′-hydroxylation phenotypes. J. Pharmacol. Exp. Ther. 1994, 271, 860–867. [Google Scholar] [PubMed]
- Koyama, E.; Chiba, K.; Tani, M.; Ishizaki, T. Reappraisal of human CYP isoforms involved in imipramine N-demethylation and 2-hydroxylation: A study using microsomes obtained from putative extensive and poor metabolizers of S-mephenytoin and eleven recombinant human CYPs. J. Pharmacol. Exp. Ther. 1997, 281, 1199–1210. [Google Scholar] [PubMed]
- Faassen, F.; Vogel, G.; Spanings, H.; Vromans, H. Caco-2 permeability, P-glycoprotein transport ratios and brain penetration of heterocyclic drugs. Int. J. Pharm. 2003, 263, 113–122. [Google Scholar] [CrossRef]
- Smith, C.J.; Ryckman, K.K.; Bahr, T.M.; Dagle, J.M. Polymorphisms in CYP2C9 are associated with response to indomethacin among neonates with patent ductus arteriosus. Pediatr. Res. 2017, 82, 776–780. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Zhou, Z.-W.; Yang, L.-P.; Cai, J.-P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef]
- Venkataraman, H.; den Braver, M.W.; Vermeulen, N.P.E.; Commandeur, J.N.M. Cytochrome P450-mediated bioactivation of mefenamic acid to quinoneimine intermediates and inactivation by human glutathione S-transferases. Chem. Res. Toxicol. 2014, 27, 2071–2081. [Google Scholar] [CrossRef]
- Levran, O.; O’Hara, K.; Peles, E.; Li, D.; Barral, S.; Ray, B.; Borg, L.; Ott, J.; Adelson, M.; Kreek, M.J. ABCB1 (MDR1) genetic variants are associated with methadone doses required for effective treatment of heroin dependence. Hum. Mol. Genet. 2008, 17, 2219–2227. [Google Scholar] [CrossRef]
- Chen, C.-H.; Wang, S.-C.; Tsou, H.-H.; Ho, I.-K.; Tian, J.-N.; Yu, C.-J.; Hsiao, C.-F.; Chou, S.-Y.; Lin, Y.-F.; Fang, K.-C.; et al. Genetic polymorphisms in CYP3A4 are associated with withdrawal symptoms and adverse reactions in methadone maintenance patients. Pharmacogenomics 2011, 12, 1397–1406. [Google Scholar] [CrossRef]
- Crist, R.C.; Li, J.; Doyle, G.A.; Gilbert, A.; Dechairo, B.M.; Berrettini, W.H. Pharmacogenetic analysis of opioid dependence treatment dose and dropout rate. Am. J. Drug Alcohol Abuse 2018, 44, 431–440. [Google Scholar] [CrossRef]
- Lan, T.; Yuan, L.-J.; Hu, X.-X.; Zhou, Q.; Wang, J.; Huang, X.-X.; Dai, D.-P.; Cai, J.-P.; Hu, G.-X. Effects of CYP2C19 variants on methadone metabolism in vitro. Drug Test Anal. 2017, 9, 634–639. [Google Scholar] [CrossRef]
- Ahmad, T.; Valentovic, M.A.; Rankin, G.O. Effects of cytochrome P450 single nucleotide polymorphisms on methadone metabolism and pharmacodynamics. Biochem. Pharmacol. 2018, 153, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Campa, D.; Gioia, A.; Tomei, A.; Poli, P.; Barale, R. Association of ABCB1/MDR1 and OPRM1 gene polymorphisms with morphine pain relief. Clin. Pharmacol. Ther. 2008, 83, 559–566. [Google Scholar] [CrossRef]
- Olesen, O.V.; Linnet, K. Hydroxylation and demethylation of the tricyclic antidepressant nortriptyline by cDNA-expressed human cytochrome P-450 isozymes. Drug Metab. Dispos. 1997, 25, 740–744. [Google Scholar]
- Venkatakrishnan, K.; von Moltke, L.L.; Greenblatt, D.J. Nortriptyline E-10-hydroxylation in vitro is mediated by human CYP2D6 (high affinity) and CYP3A4 (low affinity): Implications for interactions with enzyme-inducing drugs. J. Clin. Pharmacol. 1999, 39, 567–577. [Google Scholar] [CrossRef]
- Lalovic, B.; Phillips, B.; Risler, L.L.; Howald, W.; Shen, D.D. Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab. Dispos. 2004, 32, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.B.; Rebsamen, M.C.; Rossier, M.F.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br. J. Pharmacol. 2010, 160, 907–918. [Google Scholar] [CrossRef]
- Lötsch, J.; von Hentig, N.; Freynhagen, R.; Griessinger, N.; Zimmermann, M.; Doehring, A.; Rohrbacher, M.; Sittl, R.; Geisslinger, G. Cross-sectional analysis of the influence of currently known pharmacogenetic modulators on opioid therapy in outpatient pain centers. Pharm. Genom. 2009, 19, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef]
- Subrahmanyam, V.; Renwick, A.B.; Walters, D.G.; Young, P.J.; Price, R.J.; Tonelli, A.P.; Lake, B.G. Identification of cytochrome P-450 isoforms responsible for cis-tramadol metabolism in human liver microsomes. Drug Metab. Dispos. 2001, 29, 1146–1155. [Google Scholar]
- Paar, W.D.; Poche, S.; Gerloff, J.; Dengler, H.J. Polymorphic CYP2D6 mediates O-demethylation of the opioid analgesic tramadol. Eur. J. Clin. Pharmacol. 1997, 53, 235–239. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Müller, G.; Meineke, I.; Wernecke, K.-D.; Roots, I.; Brockmöller, J. Effects of polymorphisms in CYP2D6, CYP2C9, and CYP2C19 on trimipramine pharmacokinetics. J. Clin. Psychopharmacol. 2003, 23, 459–466. [Google Scholar] [CrossRef]
- Uhr, M.; Grauer, M.T. abcb1ab P-glycoprotein is involved in the uptake of citalopram and trimipramine into the brain of mice. J. Psychiatr. Res. 2003, 37, 179–185. [Google Scholar] [CrossRef]
- Eap, C.B.; Bender, S.; Gastpar, M.; Fischer, W.; Haarmann, C.; Powell, K.; Jonzier-Perey, M.; Cochard, N.; Baumann, P. Steady state plasma levels of the enantiomers of trimipramine and of its metabolites in CYP2D6-, CYP2C19- and CYP3A4/5-phenotyped patients. Ther. Drug. Monit. 2000, 22, 209–214. [Google Scholar] [CrossRef]
- Eap, C.B.; Lessard, E.; Baumann, P.; Brawand-Amey, M.; Yessine, M.-A.; O’Hara, G.; Turgeon, J. Role of CYP2D6 in the stereoselective disposition of venlafaxine in humans. Pharmacogenetics 2003, 13, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Uhr, M.; Tontsch, A.; Namendorf, C.; Ripke, S.; Lucae, S.; Ising, M.; Dose, T.; Ebinger, M.; Rosenhagen, M.; Kohli, M.; et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 2008, 57, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynninen, V.-V.; Olkkola, K.T.; Bertilsson, L.; Kurkinen, K.; Neuvonen, P.J.; Laine, K. Effect of terbinafine and voriconazole on the pharmacokinetics of the antidepressant venlafaxine. Clin. Pharmacol. Ther. 2008, 83, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, L.; Rollason, V.; Boehlen, F.; Rebsamen, M.; Combescure, C.; Grünenwald, M.; Matthey, A.; Bonnabry, P.; Dayer, P.; Desmeules, J.A. Impact of CYP2C9 polymorphisms on the vulnerability to pharmacokinetic drug-drug interactions during acenocoumarol treatment. Pharmacogenomics 2013, 14, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Ansermot, N.; Rebsamen, M.; Chabert, J.; Fathi, M.; Gex-Fabry, M.; Daali, Y.; Besson, M.; Rossier, M.; Rudaz, S.; Hochstrasser, D.; et al. Influence of ABCB1 gene polymorphisms and P-glycoprotein activity on cyclosporine pharmacokinetics in peripheral blood mononuclear cells in healthy volunteers. Drug Metab. Lett. 2008, 2, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Desmeules, J.; Chabert, J.; Rebsamen, M.; Rapiti, E.; Piguet, V.; Besson, M.; Dayer, P.; Cedraschi, C. Central pain sensitization, COMT Val158Met polymorphism, and emotional factors in fibromyalgia. J. Pain 2014, 15, 129–135. [Google Scholar] [CrossRef]
- PharmGKB. Available online: https://www.pharmgkb.org/ (accessed on 12 October 2018).
- Bosilkovska, M.; Samer, C.F.; Déglon, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Walder, B.; Desmeules, J.A.; Daali, Y. Geneva cocktail for cytochrome p450 and P-glycoprotein activity assessment using dried blood spots. Clin. Pharmacol. Ther. 2014, 96, 349–359. [Google Scholar] [CrossRef]
- Rebsamen, M.C.; Desmeules, J.; Daali, Y.; Chiappe, A.; Diemand, A.; Rey, C.; Chabert, J.; Dayer, P.; Hochstrasser, D.; Rossier, M.F. The AmpliChip CYP450 test: Cytochrome P450 2D6 genotype assessment and phenotype prediction. Pharm. J. 2009, 9, 34–41. [Google Scholar] [CrossRef]
- Daali, Y.; Cherkaoui, S.; Doffey-Lazeyras, F.; Dayer, P.; Desmeules, J.A. Development and validation of a chemical hydrolysis method for dextromethorphan and dextrophan determination in urine samples: Application to the assessment of CYP2D6 activity in fibromyalgia patients. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 861, 56–63. [Google Scholar] [CrossRef]
- Bosilkovska, M.; Déglon, J.; Samer, C.; Walder, B.; Desmeules, J.; Staub, C.; Daali, Y. Simultaneous LC-MS/MS quantification of P-glycoprotein and cytochrome P450 probe substrates and their metabolites in DBS and plasma. Bioanalysis 2014, 6, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daali, Y.; Samer, C.; Déglon, J.; Thomas, A.; Chabert, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Desmeules, J. Oral flurbiprofen metabolic ratio assessment using a single-point dried blood spot. Clin. Pharmacol. Ther. 2012, 91, 489–496. [Google Scholar] [CrossRef]
- Jerdi, M.C.; Daali, Y.; Oestreicher, M.K.; Cherkaoui, S.; Dayer, P. A simplified analytical method for a phenotyping cocktail of major CYP450 biotransformation routes. J. Pharm. Biomed. Anal. 2004, 35, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauschke, V.M.; Ingelman-Sundberg, M. Precision Medicine and Rare Genetic Variants. Trends Pharmacol. Sci. 2016, 37, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Crews, K.R.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Klein, T.E.; Caudle, K.E.; Haidar, C.E.; Shen, D.D.; Callaghan, J.T.; Sadhasivam, S.; et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin. Pharmacol. Ther. 2014, 95, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Rollason, V.; Samer, C.; Piguet, V.; Dayer, P.; Desmeules, J. Pharmacogenetics of analgesics: Toward the individualization of prescription. Pharmacogenomics 2008, 9, 905–933. [Google Scholar] [CrossRef]
- Knezevic, N.N.; Tverdohleb, T.; Knezevic, I.; Candido, K.D. The Role of Genetic Polymorphisms in Chronic Pain Patients. Int. J. Mol. Sci. 2018, 19, 1707. [Google Scholar] [CrossRef] [Green Version]
- Owusu Obeng, A.; Hamadeh, I.; Smith, M. Review of Opioid Pharmacogenetics and Considerations for Pain Management. Pharmacotherapy 2017, 37, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.D.; Garcia, A.J.; Hall, O.M.; Jeha, G.M.; Cramer, K.D.; Granier, A.L.; Kallurkar, A.; Cornett, E.M.; Urman, R.D. Update on the pharmacogenomics of pain management. Pharm. Pers. Med. 2019, 12, 125–143. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, P.A.; Conchon Costa, A.C.; Lauretti, G.R.; de Moraes, N.V. Pharmacogenomics in chronic pain therapy: From disease to treatment and challenges for clinical practice. Pharmacogenomics 2019, 20, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Jannetto, P.J.; Bratanow, N.C.; Clark, W.A.; Hamill-Ruth, R.J.; Hammett-Stabler, C.A.; Huestis, M.A.; Kassed, C.A.; McMillin, G.A.; Melanson, S.E.; Langman, L.J. Executive Summary: American Association of Clinical Chemistry Laboratory Medicine Practice Guideline—Using Clinical Laboratory Tests to Monitor Drug Therapy in Pain Management Patients. J. Appl. Lab. Med. 2018, 2, 489–526. [Google Scholar] [CrossRef] [Green Version]
- Vizirianakis, I.S.; Miliotou, A.N.; Mystridis, G.A.; Andriotis, E.G.; Andreadis, I.I.; Papadopoulou, L.C.; Fatouros, D.G. Tackling pharmacological response heterogeneity by PBPK modeling to advance precision medicine productivity of nanotechnology and genomics therapeutics. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 139–151. [Google Scholar] [CrossRef]
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; Van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532. [Google Scholar] [CrossRef]
| 1A2 | 2C8 | 2C9 | 2C19 | 2D6 | 3A4/5 | P-gp | |
|---|---|---|---|---|---|---|---|
| amitriptyline | ! | ! | |||||
| buprenorphine | |||||||
| celecoxib | |||||||
| clomipramine | ! | ! | |||||
| codeine | ! | ||||||
| dextromethorphan | ! | ||||||
| diclofenac | |||||||
| duloxetine | |||||||
| fentanyl | |||||||
| ibuprofen | |||||||
| imipramine | |||||||
| indomethacin | |||||||
| ketoprofen | |||||||
| mefenamic acid | |||||||
| methadone | |||||||
| morphine | |||||||
| nortriptyline | |||||||
| oxycodone | ! | ||||||
| tramadol | ! | ||||||
| trimipramine | |||||||
| venlafaxine | ! |
 ; Minor pathway
; Minor pathway  ; Active metabolite
; Active metabolite  .
.| Frequency | % | |
|---|---|---|
| Sex, female/male | 104/51 | 67.1/32.9 |
| Number of demands | ||
| 1 | 93 | 60.0 |
| 2 | 46 | 29.7 |
| 3 to 6 | 16 | 10.3 |
| Genotype assessment a | 122 | 78.7 |
| Phenotype assessment b | 78 | 50.3 |
| Analgesic drug categories (n = 243) | ||
| opioids c | 57 | 23.5 |
| Prodrug opioids d | 148 | 60.9 |
| Nonsteroidal anti-inflammatory drugs e | 15 | 6.2 |
| antidepressants f | 22 | 9.1 |
| paracetamol | 1 | 0.4 |
| Reasons for demands (n = 243) | ||
| Adverse events | 145 | 59.7 |
| Non-response | 92 | 37.9 |
| Both | 6 | 2.5 |
| Genotype | Predicted Phenotype | Measured Phenotype | |||
|---|---|---|---|---|---|
| Frequency | % | Frequency | % | ||
| CYP2D6 | n = 105 | n = 73 | |||
| UM | *1/*2xN | 3 | 2.9 | 15 | 20.6 |
| NM | *1/*1, *1/*2, *1/*3, *1/*4, *1/*5, *1/*6, *1/*10, *1/*35, *1/*41, *1XN/*4, *2/*2, *2/*4, *2/*5, *2/*6, *2/*10, *2xN/*4, *4/*35, *10/*35, *35/*35, *35/*41 | 84 | 80.0 | 27 | 37.0 |
| IM | *4/*9, *4/*41, *4/*10XN, *4XN/*41, *5/*41, *9/*41, *10/*41, *17/*41 | 11 | 10.5 | 19 | 26.0 |
| PM | *4/*4, *4/*5 | 7 | 6.7 | 12 | 16.4 |
| CYP2C9 | n = 23 | n = 27 | |||
| increased | 0 | 0.0 | 5 | 18.5 | |
| normal | *1/*1 | 15 | 65.2 | 18 | 66.7 |
| reduced | *1/*2, *1/*3 | 8 | 34.8 | 4 | 14.8 |
| CYP2C19 | n = 37 | n = 27 | |||
| UM | *17/*17 | 1 | 2.7 | 2 | 7.4 |
| NM | *1/*1 | 25 | 67.6 | 21 | 77.8 |
| IM | *1/*2, *2/*17 | 10 | 27.0 | 2 | 7.4 |
| PM | *2/*2 | 1 | 2.7 | 2 | 7.4 |
| CYP1A2 | n = 21 | ||||
| increased | 10 | 47.6 | |||
| normal | 11 | 52.4 | |||
| reduced | 0 | 0.0 | |||
| CYP3A | n = 32 | ||||
| increased | 4 | 12.5 | |||
| normal | 24 | 75.0 | |||
| reduced | 4 | 12.5 | |||
| ABCB1 C3435T (rs1045642) | n = 56 | ||||
| normal | CC | 13 | 23.2 | ||
| reduced | CT | 30 | 53.6 | ||
| reduced | TT | 13 | 23.2 | ||
| ABCB1 G2677T (rs2032582) | n = 54 | ||||
| normal | GG | 18 | 33.3 | ||
| reduced | GT/GA | 28 | 51.9 | ||
| reduced | TT | 8 | 14.8 | ||
| COMT rs4680 | n = 33 | ||||
| normal | GG | 4 | 12.1 | ||
| reduced | GA | 21 | 63.6 | ||
| reduced | AA | 8 | 24.2 | ||
| Predicted Phenotype | n | % Concordance a | Measured Phenotype | Co-medication Possibly Relevant to Discordant Cases (Predicted > Measured Phenotype) | |||
|---|---|---|---|---|---|---|---|
| UM Frequency | NM Frequency | IM Frequency | PM Frequency | ||||
| CYP2D6 | 40 | 52.5 | |||||
| UM | 0 | 0 | 1 | 0 | citalopram (UM > IM) | ||
| NM | 9 | 14 | 7 | 1 | fluoxetine (NM > IM); citalopram (NM > IM); duloxetine (NM > IM); multiple CYP2D6 substrates and phytotherapy (NM > IM) | ||
| IM | 0 | 0 | 6 | 0 | |||
| PM | 0 | 1 | 0 | 1 | |||
| CYP2C9 | 8 | 50.0 | |||||
| NM | 2 | 3 | 0 | 0 | |||
| IM | 0 | 2 | 1 | 0 | |||
| CYP2C19 | 5 | 100.0 | |||||
| NM | 0 | 5 | 0 | 0 | |||
| Type of Demand | Linkage with Genotype or Phenotype | Examples | ||
|---|---|---|---|---|
| Estimated Probability a | Frequency | % | ||
| Adverse events | ||||
| All drugs (n = 145) | intermediate | 30 | 20.7 | - |
| high | 28 | 19.3 | - | |
| Opioids (n = 43) | intermediate | 14 | 32.6 | Marked drowsiness for 24 h with buprenorphine; homozygous mutation for ABCB1 C3435T: TT and heterozygous mutation for G2677T/A: GT |
| high | 2 | 4.7 | Prolonged altered consciousness state with fentanyl; CY3A4 PM phenotype | |
| Prodrug opioids (n = 81) | intermediate | 13 | 16.0 | Malaise with dextromethorphan; CYP2D6 IM genotype and phenotype |
| high | 24 | 29.6 | Severe vomiting with codeine; CYP2D6 UM phenotype | |
| NSAIDs (n = 5) | intermediate | 2 | 40.0 | Numerous adverse reactions with diclofenac; CYP2C9 IM genotype |
| high | 0 | 0.0 | - | |
| Antidepressants (n = 16) | intermediate | 1 | 6.3 | Numerous adverse reactions with imipramine; CYP2D6 IM genotype and phenotype |
| high | 2 | 12.5 | Drowsiness and confusion with trimipramine; CYP2D6 PM phenotype | |
| Non-response | ||||
| All drugs (n = 92) | intermediate | 12 | 13.0 | - |
| high | 14 | 15.2 | - | |
| Opioids (n = 11) | intermediate | 0 | 0.0 | - |
| high | 0 | 0.0 | - | |
| Prodrug opioids (n = 64) | intermediate | 12 | 18.8 | Non-response to oxycodone |
| high | 11 | 17.2 | Non-response to codeine; CYP2D6 PM phenotype | |
| NSAIDs (n = 10) | intermediate | 0 | 0.0 | - |
| high | 1 | 10.0 | Non-response to ibuprofen; CYP2C9 UM phenotype | |
| Antidepressants (n = 6) | intermediate | 0 | 0.0 | - |
| high | 1 | 16.7 | Non-response to amitriptyline; CYP2D6 PM phenotype | |
| Type of Demand | Linkage with Genotype or Phenotype | Examples | ||
|---|---|---|---|---|
| Estimated Probability a | Frequency | % | ||
| Adverse events | ||||
| All drugs (n = 145) | intermediate | 30 | 20.7 | - |
| high | 28 | 19.3 | - | |
| Tramadol (n = 49) | intermediate | 7 | 14.3 | Sedation, dizziness; CYP2D6 IM genotype and phenotype |
| high | 12 | 24.5 | Sedation and hallucinations; CYP2D6 UM genotype | |
| Morphine (n = 32) | intermediate | 12 | 37.5 | Comateous state; homozygous mutation for COMT rs 4680 A/A |
| high | 0 | 0.0 | - | |
| Codeine (n = 19) | intermediate | 4 | 21.1 | Hallucination; CYP2D6 IM genotype |
| high | 9 | 47.4 | Important drowsiness; CYP2D6 UM phenotype | |
| Oxycodone (n = 7) | intermediate | 1 | 14.3 | Drowsiness; CYP2D6 IM genotype |
| high | 1 | 14.3 | Disorientation and sedation; CYP2D6 UM phenotype | |
| Non-response | ||||
| All drugs (n = 92) | intermediate | 12 | 13.0 | - |
| high | 14 | 15.2 | - | |
| Tramadol (n = 33) | intermediate | 7 | 21.2 | Non-response; CYP2D6 IM phenotype |
| high | 6 | 18.2 | Non-response; CYP2D6 PM phenotype | |
| Morphine (n = 4) | intermediate | 0 | 0.0 | - |
| high | 0 | 0.0 | - | |
| Codeine (n = 13) | intermediate | 4 | 30.8 | Non-response; CYP2D6 IM phenotype |
| high | 1 | 7.7 | Non-response; CYP2D6 PM phenotype | |
| Oxycodone (n = 17) | intermediate | 1 | 5.9 | Non-response; CYP2D6 IM phenotype |
| high | 4 | 23.5 | Non-response; CYP2D6 PM phenotype | |
| Drug | Type of Demand | n | Measured CYP2D6 Phenotype | p-Value a | |||
|---|---|---|---|---|---|---|---|
| UM Frequency (%) | NM Frequency (%) | IM Frequency (%) | PM Frequency (%) | ||||
| Tramadol | 0.50 | ||||||
| Adverse events | 24 | 6 (25.0) | 9 (37.5) | 6 (25.0) | 3 (12.5) | ||
| Non-response | 15 | 1 (6.7) | 6 (40.0) | 6 (40.0) | 2 (13.3) | ||
| Codeine | 0.071 | ||||||
| Adverse events | 13 | 5 (38.5) | 6 (46.2) | 1 (7.7) | 1 (7.7) | ||
| Non-response | 9 | 0 (0.0) | 4 (44.4) | 4 (44.4) | 1 (11.1) | ||
| Oxycodone | 0.78 | ||||||
| Adverse events | 3 | 1 (33.3) | 2 (66.7) | 0 (0.0) | 0 (0.0) | ||
| Non-response | 9 | 3 (33.3) | 2 (22.2) | 1 (11.1) | 3 (33.3) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rollason, V.; Lloret-Linares, C.; Lorenzini, K.I.; Daali, Y.; Gex-Fabry, M.; Piguet, V.; Besson, M.; Samer, C.; Desmeules, J. Evaluation of Phenotypic and Genotypic Variations of Drug Metabolising Enzymes and Transporters in Chronic Pain Patients Facing Adverse Drug Reactions or Non-Response to Analgesics: A Retrospective Study. J. Pers. Med. 2020, 10, 198. https://doi.org/10.3390/jpm10040198
Rollason V, Lloret-Linares C, Lorenzini KI, Daali Y, Gex-Fabry M, Piguet V, Besson M, Samer C, Desmeules J. Evaluation of Phenotypic and Genotypic Variations of Drug Metabolising Enzymes and Transporters in Chronic Pain Patients Facing Adverse Drug Reactions or Non-Response to Analgesics: A Retrospective Study. Journal of Personalized Medicine. 2020; 10(4):198. https://doi.org/10.3390/jpm10040198
Chicago/Turabian StyleRollason, Victoria, Célia Lloret-Linares, Kuntheavy Ing Lorenzini, Youssef Daali, Marianne Gex-Fabry, Valérie Piguet, Marie Besson, Caroline Samer, and Jules Desmeules. 2020. "Evaluation of Phenotypic and Genotypic Variations of Drug Metabolising Enzymes and Transporters in Chronic Pain Patients Facing Adverse Drug Reactions or Non-Response to Analgesics: A Retrospective Study" Journal of Personalized Medicine 10, no. 4: 198. https://doi.org/10.3390/jpm10040198