
Novel KLK4 Mutations Cause Hypomaturation Amelogenesis Imperfecta

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment of Study Subjects
2.2. DNA Isolation and Whole Exome Sequencing
2.3. Analysis of the Sequencing Reads
2.4. Polymerase Chain Reaction (PCR) and Sanger Sequencing
2.5. PCR Mutagenesis
2.6. Western Blotting
2.7. Zymography
3. Results
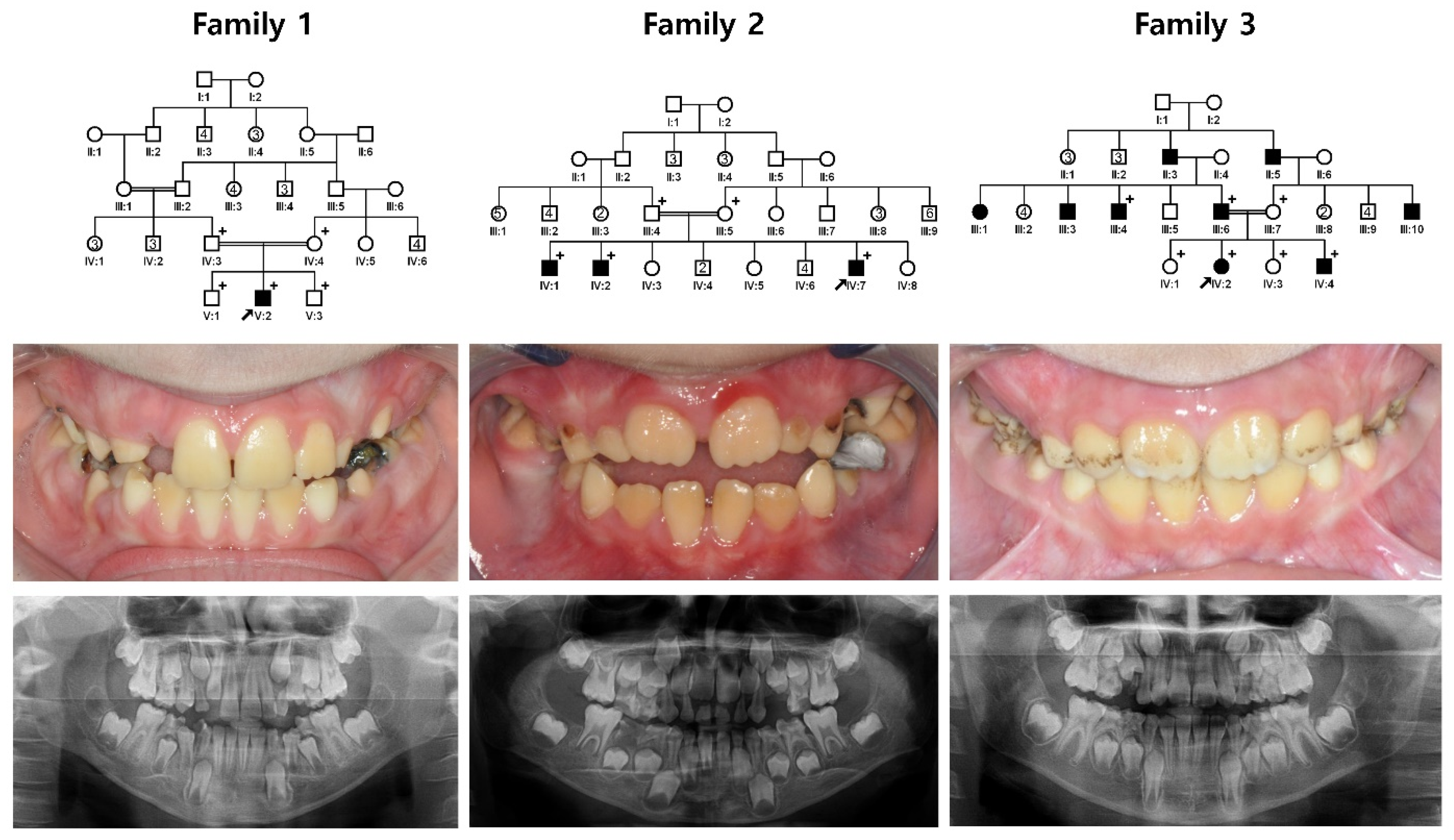
3.1. Family 1~3
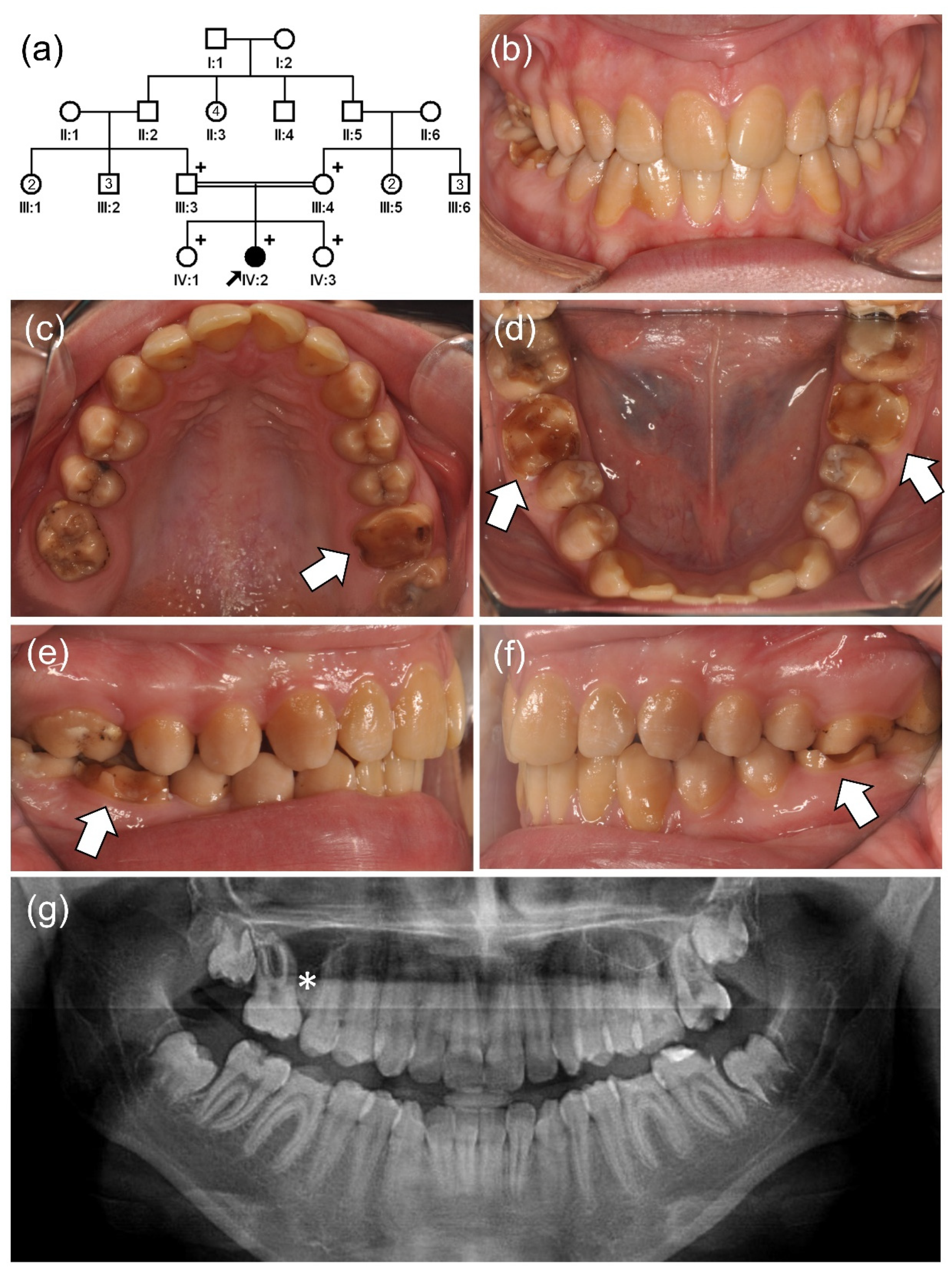
3.2. Family 4
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simmer, J.P.; Papagerakis, P.; Smith, C.E.; Fisher, D.C.; Rountrey, A.; Zheng, L.; Hu, J.C.-C. Regulation of Dental Enamel Shape and Hardness. J. Dent. Res. 2010, 89, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Salanitri, S.; Seow, W.K. Developmental enamel defects in the primary dentition: Aetiology and clinical management. Aust. Dent. J. 2013, 58, 133–140, quiz 266. [Google Scholar] [CrossRef] [PubMed]
- Witkop, C.J., Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classification. J. Oral Pathol. Med. 1988, 17, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Carrion, I.A.; Morris, C. The Molecular Basis of Hereditary Enamel Defects in Humans. J. Dent. Res. 2015, 94, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thesleff, I. Differentiation of ameloblasts and its regulation of epithelial-mesenchymal interactions. In Dental Enamel Formation to Destruction; Robinson, C., Kirkham, J., Shore, R., Eds.; CRC Press: Boca Raton, FL, USA, 1995; p. 272. [Google Scholar]
- Wright, J.T. The molecular etiologies and associated phenotypes of amelogenesis imperfecta. Am. J. Med. Genet. Part A 2006, 140A, 2547–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, M.K.; Laouina, S.; El Alloussi, M.; Dollfus, H.; Bloch-Zupan, A. Amelogenesis Imperfecta: 1 Family, 2 Phenotypes, and 2 Mutated Genes. J. Dent. Res. 2016, 95, 1457–1463. [Google Scholar] [CrossRef]
- Parry, D.; Brookes, S.; Logan, C.; Poulter, J.; El-Sayed, W.; Al-Bahlani, S.; Al Harasi, S.; Sayed, J.; Raïf, E.M.; Shore, R.C.; et al. Mutations in C4orf26, Encoding a Peptide with In Vitro Hydroxyapatite Crystal Nucleation and Growth Activity, Cause Amelogenesis Imperfecta. Am. J. Hum. Genet. 2012, 91, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Simmer, J.P.; Hart, T.C.; Hart, P.S.; Ramaswami, M.D.; Bartlett, J.D.; Hu, J.C.-C. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J. Med. Genet. 2005, 42, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Hart, P.S.; Hart, T.C.; Michalec, M.D.; Ryu, O.H.; Simmons, D.; Hong, S.; Wright, J.T. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J. Med. Genet. 2004, 41, 545–549. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed, W.; Parry, D.A.; Shore, R.C.; Ahmed, M.; Jafri, H.; Rashid, Y.; Al-Bahlani, S.; Al Harasi, S.; Kirkham, J.; Inglehearn, C.F.; et al. Mutations in the Beta Propeller WDR72 Cause Autosomal-Recessive Hypomaturation Amelogenesis Imperfecta. Am. J. Hum. Genet. 2009, 85, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.; Poulter, J.; Logan, C.; Brookes, S.J.; Jafri, H.; Ferguson, C.H.; Anwari, B.M.; Rashid, Y.; Zhao, H.; Johnson, C.A.; et al. Identification of Mutations in SLC24A4, Encoding a Potassium-Dependent Sodium/Calcium Exchanger, as a Cause of Amelogenesis Imperfecta. Am. J. Hum. Genet. 2013, 92, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.A.; Smith, C.E.; El-Sayed, W.; Poulter, J.A.; Shore, R.C.; Logan, C.V.; Mogi, C.; Sato, K.; Okajima, F.; Harada, A.; et al. Mutations in the pH-Sensing G-protein-Coupled Receptor GPR68 Cause Amelogenesis Imperfecta. Am. J. Hum. Genet. 2016, 99, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.P.; Luder, H.; Simmons, D.; Daly, B.; Sukarawan, W.; Wright, J.T. DLX3 mutation leads to a mild phenotype of tricho-dento-osseous syndrome. J. Dent. Res. 2006, 85, 1035. [Google Scholar]
- Lee, S.K.; Lee, Z.H.; Lee, S.J.; Ahn, B.D.; Kim, Y.J.; Lee, S.H.; Kim, J.W. DLX3 mutation in a new family and its phenotypic variations. J. Dent. Res. 2008, 87, 354–357. [Google Scholar] [CrossRef]
- Ravassipour, D.; Hart, P.; Hart, T.; Ritter, A.; Yamauchi, M.; Gibson, C.; Wright, J. Unique Enamel Phenotype Associated with Amelogenin Gene (AMELX) Codon 41 Point Mutation. J. Dent. Res. 2000, 79, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Hart, P.S.; Aldred, M.J.; Seow, K.; Crawford, P.J.; Hong, S.P.; Gibson, C.W.; Hart, T.C. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect. Tissue Res. 2003, 44 (Suppl. 1), 72–78. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Seymen, F.; Park, J.-C.; Lee, K.-E.; Lee, H.K.; Lee, D.-S.; Koruyucu, M.; Gencay, K.; Bayram, M.; Tuna, E.; Lee, Z.; et al. Novel MMP20 and KLK4 Mutations in Amelogenesis Imperfecta. J. Dent. Res. 2015, 94, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Pearce, D.A. Nonsense-mediated decay in genetic disease: Friend or foe? Mutat. Res. Rev. Mutat. Res. 2014, 762, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-K.; Hu, Y.; Simmer, J.; Seymen, F.; Estrella, N.; Pal, S.; Reid, B.; Yildirim, M.; Bayram, M.; Bartlett, J.; et al. Novel KLK4 and MMP20 Mutations Discovered by Whole-exome Sequencing. J. Dent. Res. 2013, 92, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.E.L.; Kirkham, J.; Day, P.F.; Soldani, F.; McDerra, E.J.; Poulter, J.; Inglehearn, C.F.; Mighell, A.J.; Brookes, S. A Fourth KLK4 Mutation Is Associated with Enamel Hypomineralisation and Structural Abnormalities. Front. Physiol. 2017, 8, 333. [Google Scholar] [CrossRef] [Green Version]
- Pathak, M.; Wong, S.S.; Dreveny, I.; Emsley, J. Structure of plasma and tissue kallikreins. Thromb. Haemost. 2013, 110, 423–433. [Google Scholar] [CrossRef]
- Lundwall, Å.; Brattsand, M. Kallikrein-related peptidases. Cell. Mol. Life Sci. 2008, 65, 2019–2038. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Hu, J.C.-C.; Simmer, J.P. Evolution of Klk4 and enamel maturation in eutherians. Biol. Chem. 2014, 395, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, G.M.; Diamandis, E.P. The New Human Tissue Kallikrein Gene Family: Structure, Function, and Association to Disease. Endocr. Rev. 2001, 22, 184–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, J.L.; Bartlett, J.D.; Chaparian, M.G.; Fukae, M.; Uchida, T.; Xue, J.; Hu, C.-C.; Simmer, J.P. Enamel Matrix Serine Proteinase 1: Stage-Specific Expression and Molecular Modeling. Connect. Tissue Res. 1998, 39, 111–122. [Google Scholar] [CrossRef]
- Katz, B.A.; Liu, B.; Barnes, M.; Springman, E.B. Crystal structure of recombinant human tissue kallikrein at 2.0 Å resolution. Protein Sci. 1998, 7, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Debela, M.; Magdolen, V.; Grimminger, V.; Sommerhoff, C.; Messerschmidt, A.; Huber, R.; Friedrich, R.; Bode, W.; Goettig, P. Crystal Structures of Human Tissue Kallikrein 4: Activity Modulation by a Specific Zinc Binding Site. J. Mol. Biol. 2006, 362, 1094–1107. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, J.C.-C.; Smith, C.E.; Bartlett, J.D.; Simmer, J.P. Kallikrein-related peptidase 4, matrix metalloproteinase 20, and the maturation of murine and porcine enamel. Eur. J. Oral Sci. 2011, 119, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Debela, M.; Beaufort, N.; Magdolen, V.; Schechter, N.M.; Craik, C.S.; Schmitt, M.; Bode, W.; Goettig, P. Structures and specificity of the human kallikrein-related peptidases KLK 4, 5, 6, and 7. Biol. Chem. 2008, 389, 623–632. [Google Scholar] [CrossRef]
- Ryu, O.; Hu, J.C.-C.; Yamakoshi, Y.; Villemain, J.L.; Cao, X.; Zhang, C.; Bartlett, J.D.; Simmer, J.P. Porcine kallikrein-4 activation, glycosylation, activity, and expression in prokaryotic and eukaryotic hosts. Eur. J. Oral Sci. 2002, 110, 358–365. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Richardson, A.S.; Nunez, S.M.; Yamakoshi, F.; Milkovich, R.N.; Hu, J.C.-C.; Bartlett, J.D.; Simmer, J.P. Enamel proteins and proteases in Mmp20 and Klk4 null and double-null mice. Eur. J. Oral Sci. 2011, 119 (Suppl. 1), 206–216. [Google Scholar] [CrossRef] [Green Version]
- Yamakoshi, Y.; Simmer, J.P.; Bartlett, J.D.; Karakida, T.; Oida, S. MMP20 and KLK4 activation and inactivation interactions in vitro. Arch. Oral Biol. 2013, 58, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Yamakoshi, Y.; Yamakoshi, F.; Hu, J.C.-C.; Simmer, J.P. Characterization of kallikrein-related peptidase 4 glycosylations. Eur. J. Oral Sci. 2011, 119, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, J.D.; Simmer, J.P. Kallikrein-related peptidase-4 (KLK4): Role in enamel formation and revelations from ablated mice. Front. Physiol. 2014, 5, 240. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | cDNA | Protein | Mode of Inheritance | References |
|---|---|---|---|---|
| Exon 3 | c.170C>A | p.(Ser57*) | AR homo | This report |
| Exon 4 | c.245delG | p.(Gly82Alafs*87) | AR homo | Wang et al. (2013) [28] |
| Exon 4 | c.458G>A | p.(Trp153*) | AR homo | Hart et al. (2004) [10] |
| Exon 6 | c.620_621delCT | p.(Ser207Trpfs*38) | AR homo | Seymen et al. (2015) [22] |
| Exon 6 | c.632delT | p.(Leu211Argfs*37) | AR homo | Smith et al. (2017) [29] |
| Exon 6 | c.637T>C | p.(Cys213Arg) | AR homo | This report |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.; Zhang, H.; Seymen, F.; Kim, Y.J.; Kasimoglu, Y.; Koruyucu, M.; Simmer, J.P.; Hu, J.C.-C.; Kim, J.-W. Novel KLK4 Mutations Cause Hypomaturation Amelogenesis Imperfecta. J. Pers. Med. 2022, 12, 150. https://doi.org/10.3390/jpm12020150
Lee Y, Zhang H, Seymen F, Kim YJ, Kasimoglu Y, Koruyucu M, Simmer JP, Hu JC-C, Kim J-W. Novel KLK4 Mutations Cause Hypomaturation Amelogenesis Imperfecta. Journal of Personalized Medicine. 2022; 12(2):150. https://doi.org/10.3390/jpm12020150
Chicago/Turabian StyleLee, Yejin, Hong Zhang, Figen Seymen, Youn Jung Kim, Yelda Kasimoglu, Mine Koruyucu, James P. Simmer, Jan C.-C. Hu, and Jung-Wook Kim. 2022. "Novel KLK4 Mutations Cause Hypomaturation Amelogenesis Imperfecta" Journal of Personalized Medicine 12, no. 2: 150. https://doi.org/10.3390/jpm12020150