
Population Genetics for Inferring Introduction Sources of the Oriental Fruit Fly, Bactrocera dorsalis: A Test for Quarantine Use in Korea

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon, Sample Collection and DNA Extraction
2.2. Haplotype Network Analysis
2.3. Population Genetics Analysis
2.3.1. Microsatellite Marker Screening and Design of Multiplex PCR Set
2.3.2. Multiplex PCR and Fragment Analysis
2.3.3. Data Analysis
3. Results
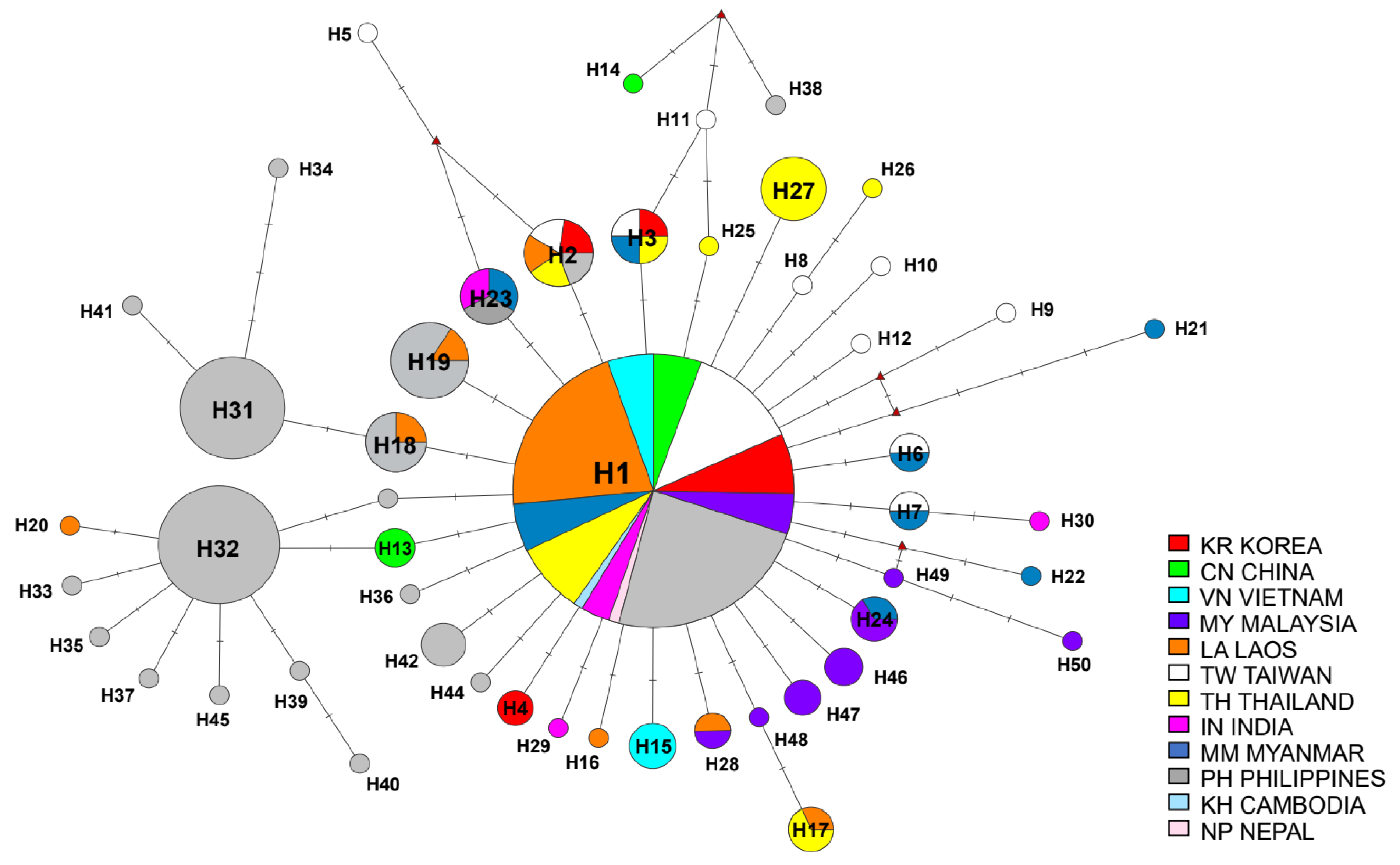
3.1. Haplotype Network
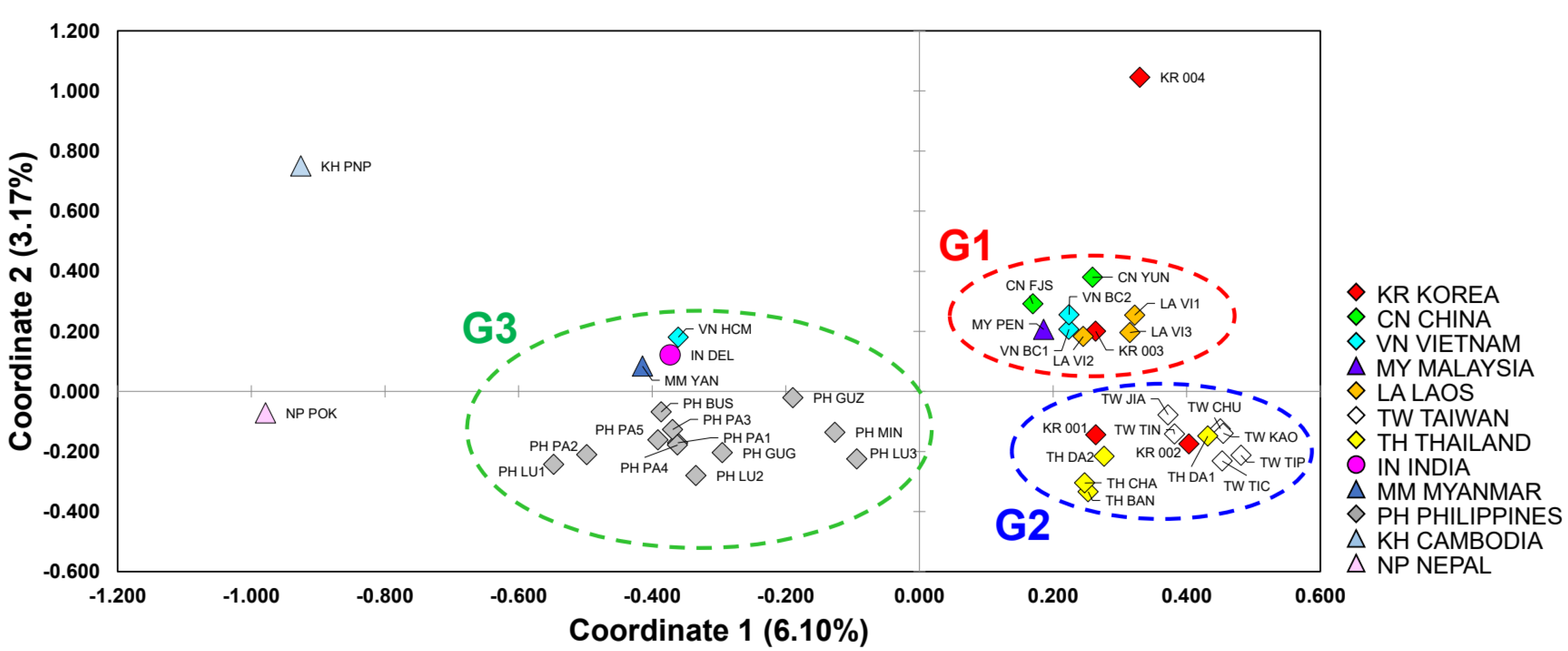
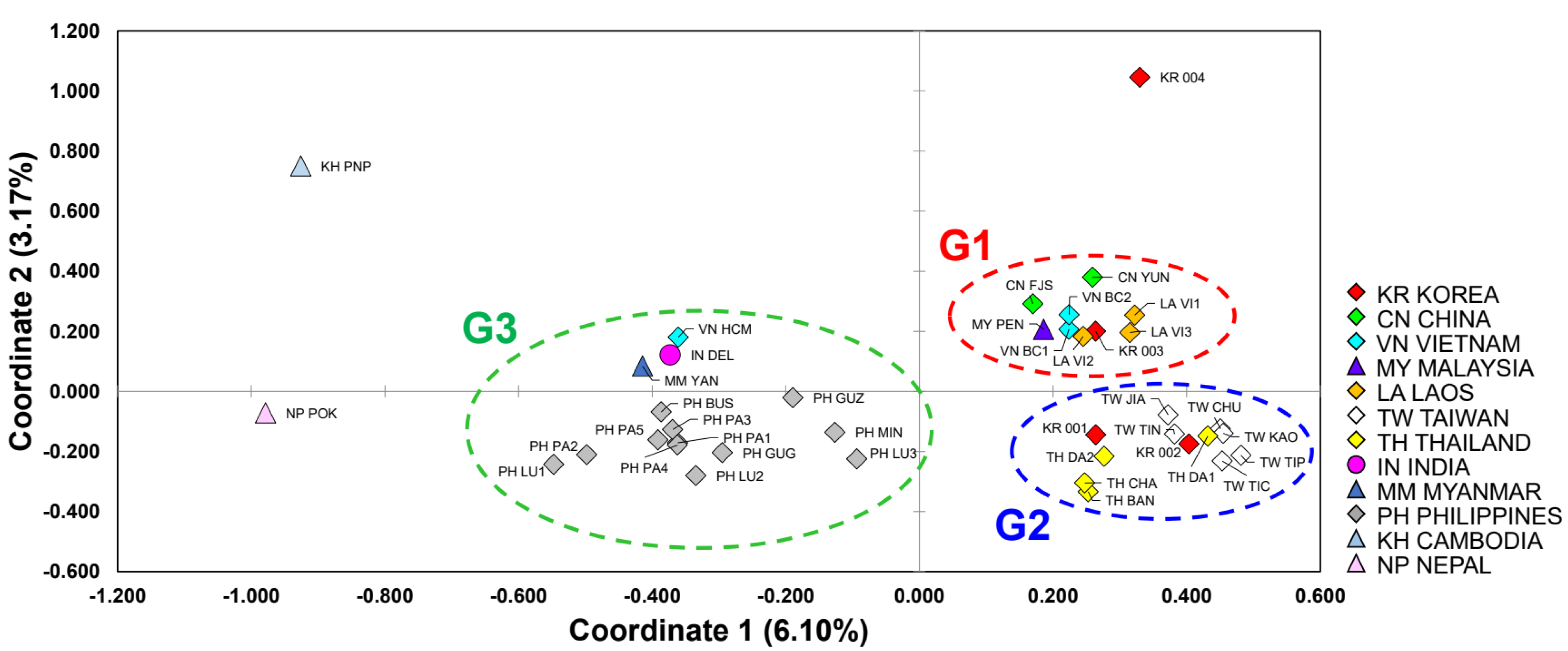
3.2. Genetic Differentiation within and between Populations
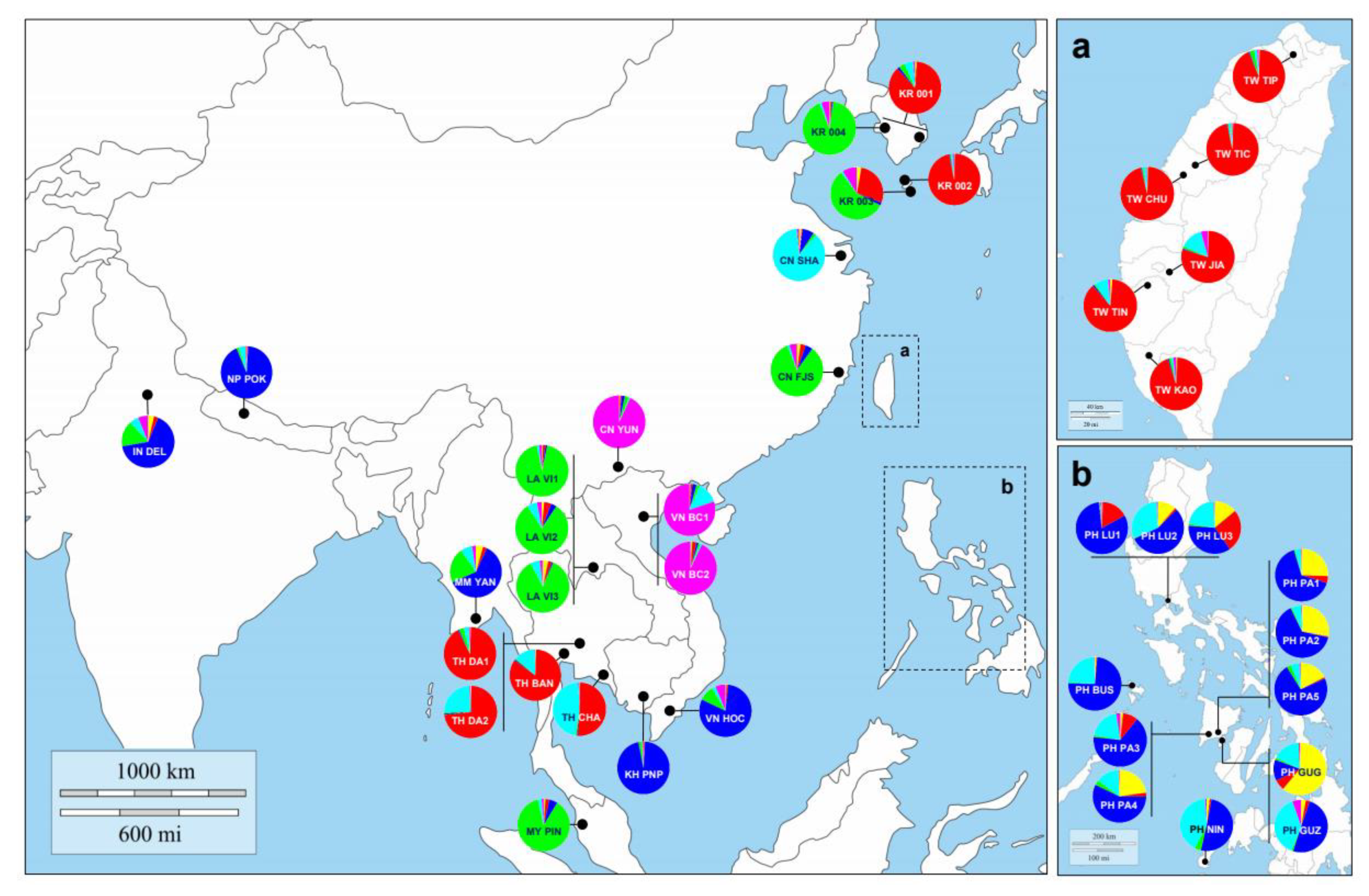
3.3. Genetic Structure and Assignment
3.4. Inferring an Introduction to Test Hypothetical Scenarios by ABC Analysis
4. Discussion
4.1. The Genetic Structure and the Global Origin of Bactrocera dorsalis
4.2. Inferring Source Population for Korean Quarantine Samples
4.3. Applications for Future Quarantine
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vanhanen, H. Invasive insects in Europe-the role of climate change and global trade. Diss. For. 2008, 57, 33. [Google Scholar] [CrossRef] [Green Version]
- Bale, J.; Masters, G.J.; Hodkinson, I.; Awmack, C.; Bezemer, T.M.; Brown, V.K.; Butterfield, J.; Buse, A.; Coulson, J.C.; Farrar, J.; et al. Herbivory in global climate change research: Direct effects of rising temperature on insect herbivores. Glob. Chang. Biol. 2002, 8, 1–16. [Google Scholar] [CrossRef]
- Gippet, J.M.; Liebhold, A.M.; Fenn-Moltu, G.; Bertelsmeier, C. Human-mediated dispersal in insects. Curr. Opin. Insect Sci. 2019, 35, 96–102. [Google Scholar] [CrossRef]
- Roques, A. Alien forest insects in a warmer world and a globalised economy: Impacts of changes in trade, tourism and climate on forest biosecurity. N. Z. J. For. Sci. 2010, 40, 77–94. [Google Scholar]
- Kwon, Y.-S.; Chung, N.; Bae, M.-J.; Li, F.; Chon, T.-S.; Park, Y.-S. Effects of meteorological factors and global warming on rice insect pests in Korea. J. Asia-Pac. Entomol. 2012, 15, 507–515. [Google Scholar] [CrossRef]
- Seebens, H.; Blackburn, T.M.; Dyer, E.E.; Genovesi, P.; Hulme, P.E.; Jeschke, J.M.; Pagad, S.; Pyšek, P.; Winter, M.; Arianoutsou, M. No saturation in the accumulation of alien species worldwide. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-A.; Kwon, W.-T.; Boo, K.-O. Future projections on the change of onset date and duration of natural seasons using SRES A1B data in South Korea. J. Korean Geogr. Soc. 2007, 42, 835–850. [Google Scholar]
- Pyšek, P.; Richardson, D.M. Invasive species, environmental change and management, and health. Annu. Rev. Environ. Resour. 2010, 35, 25–55. [Google Scholar] [CrossRef] [Green Version]
- EPPO. Bactrocera dorsalis. In EPPO/CABI Quarantine Pests for Europe, 2nd ed.; Smith, I.M., McNamara, D.G., Scott, P.R., Holderness, M., Eds.; EPPO: Paris, France, 1997. [Google Scholar]
- Clarke, A.R.; Armstrong, K.F.; Carmichael, A.E.; Milne, J.R.; Raghu, S.; Roderick, G.K.; Yeates, D.K. Invasive phytophagous pests arising through a recent tropical evolutionary radiation: The Bactrocera dorsalis complex of fruit flies. Annu. Rev. Entomol. 2005, 50, 293–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, R.I.; Piñero, J.C.; Leblanc, L. An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific Region. Insects 2015, 6, 297–318. [Google Scholar] [CrossRef] [Green Version]
- Boykin, L.; Schutze, M.; Krosch, M.; Chomič, A.; Chapman, T.; Englezou, A.; Armstrong, K.; Clarke, A.; Hailstones, D.; Cameron, S. Multi-gene phylogenetic analysis of south-east Asian pest members of the Bactrocera dorsalis species complex (Diptera: Tephritidae) does not support current taxonomy. J. Appl. Entomol. 2014, 138, 235–253. [Google Scholar] [CrossRef]
- Schutze, M.K.; Aketarawong, N.; Amornsak, W.; Armstrong, K.F.; Augustinos, A.A.; Barr, N.; Bo, W.; Bourtzis, K.; Boykin, L.M.; Caceres, C. Synonymization of key pest species within the Bactrocera dorsalis species complex (Diptera: Tephritidae): Taxonomic changes based on a review of 20 years of integrative morphological, molecular, cytogenetic, behavioural and chemoecological data. Syst. Entomol. 2015, 40, 456–471. [Google Scholar] [CrossRef]
- Stephens, A.; Kriticos, D.J.; Leriche, A. The current and future potential geographical distribution of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). Bull. Entomol. Res. 2007, 97, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, N.; Kuba, H.; Soemori, H. Mating behavior of the oriental fruit fly, Dacus dorsalis Hendel (Diptera: Tephritidae). Appl. Entomol. Zool. 1984, 19, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Drew, R.A.; Hancock, D.L. The Bactrocera dorsalis complex of fruit flies (Diptera: Tephritidae: Dacinae) in Asia. Bull. Entomol. Res. Suppl. Ser. 1994, 2, 1–68. [Google Scholar] [CrossRef]
- Liquido, N.J. Effect of ripeness and location of papaya fruits on the parasitization rates of oriental fruit fly and melon fly (Diptera: Tephritidae) by braconid (Hymenoptera) parasitoids. Environ. Entomol. 1991, 20, 1732–1736. [Google Scholar] [CrossRef]
- Vargas, R.I.; Walsh, W.A.; Kanehisa, D.; Jang, E.B.; Armstrong, J.W. Demography of four Hawaiian fruit flies (Diptera: Tephritidae) reared at five constant temperatures. Ann. Entomol. Soc. Am. 1997, 90, 162–168. [Google Scholar] [CrossRef]
- De Villiers, M.; Hattingh, V.; Kriticos, D.J.; Brunel, S.; Vayssières, J.-F.; Sinzogan, A.; Billah, M.; Mohamed, S.; Mwatawala, M.; Abdelgader, H. The potential distribution of Bactrocera dorsalis: Considering phenology and irrigation patterns. Bull. Entomol. Res. 2016, 106, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Wang, X.; Niu, C.-Y.; Dong, Y.-C.; Zhu, J.-Q.; Desneux, N. Population dynamics, phenology, and overwintering of Bactrocera dorsalis (Diptera: Tephritidae) in Hubei Province, China. J. Pest Sci. 2011, 84, 289–295. [Google Scholar] [CrossRef]
- Chung, Y.-S.; Yoon, M.-B.; Kim, H.-S. On climate variations and changes observed in South Korea. Clim. Change 2004, 66, 151–161. [Google Scholar] [CrossRef]
- Boo, K.O.; Kwon, W.T.; Baek, H.J. Change of extreme events of temperature and precipitation over Korea using regional projection of future climate change. Geophys. Res. Lett. 2006, 33, 128522728. [Google Scholar] [CrossRef]
- Oh, S.-N.; Kim, Y.-H.; Hyun, M.-S. Impact of urbanization on climate change in Korea, 1973–2002. Asia-Pac. J. Atmos. Sci. 2004, 40, 725–740. [Google Scholar]
- Hulme, M.; Zhao, Z.C.; Jiang, T. Recent and future climate change in East Asia. Int. J. Climatol. 1994, 14, 637–658. [Google Scholar] [CrossRef]
- Gong, D.-Y.; Ho, C.-H. The Siberian High and climate change over middle to high latitude Asia. Theor. Appl. Climatol. 2002, 72, 1–9. [Google Scholar] [CrossRef]
- Nicholls, R.J. Coastal megacities and climate change. GeoJournal 1995, 37, 369–379. [Google Scholar] [CrossRef]
- Liang, F.; Wu, J.; Liang, G. The first report of the test on the flight ability of oriental fruit fly. Acta Agriculturae Universitatis Jiangxiensis 2001, 23, 259–260. [Google Scholar]
- Wan, X.; Liu, Y.; Zhang, B. Invasion history of the oriental fruit fly, Bactrocera dorsalis, in the Pacific-Asia region: Two main invasion routes. PLoS ONE 2012, 7, e36176. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, N.T. Fruit fly invasion: Historical, biological, economic aspects and management. In Trapping and the Detection, Control, and Regulation of Tephritid Fruit Flies; Springer: Berlin/Heidelberg, Germany, 2014; pp. 219–252. [Google Scholar]
- Wu, Z.Z.; Li, H.M.; Bin, S.Y.; Ma, J.; He, H.L.; Li, X.F.; Gong, F.L.; Lin, J.T. Sequence analysis of mitochondrial ND1 gene can reveal the genetic structure and origin of Bactrocera dorsalis ss. BMC Evol. Biol. 2014, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-j. The fruit flies of the East Asian region. Acta Zootaxonomica Sinica Suppl. 1996, 21, 1–338. [Google Scholar]
- Weifeng, L.; Lang, Y.; Kan, T.; Ling, Z.; Guangwen, L. Microsatellite polymorphism of Bactrocera dorsalis (Hendel) populations in China. Acta Entomol. Sin. 2007, 50, 1255–1262. [Google Scholar]
- Aketarawong, N.; Bonizzoni, M.; Thanaphum, S.; Gomulski, L.M.; Gasperi, G.; Malacrida, A.R.; Gugliemino, C.R. Inferences on the population structure and colonization process of the invasive oriental fruit fly, Bactrocera dorsalis (Hendel). Mol. Ecol. 2007, 16, 3522–3532. [Google Scholar] [CrossRef]
- Shi, W.; Kerdelhue, C.; Ye, H. Population genetics of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae), in Yunnan (China) based on mitochondrial DNA sequences. Environ. Entomol. 2005, 34, 977–983. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Chen, H.; Wu, J.; Li, Z. Population structure and colonization of Bactrocera dorsalis (Diptera: Tephritidae) in China, inferred from mtDNA COI sequences. J. Appl. Entomol. 2012, 136, 241–251. [Google Scholar] [CrossRef]
- Chen, P.; Ye, H.; Mu, Q. Migration and dispersal of the oriental fruit fly, Bactrocera dorsalis in regions of Nujiang River based on fluorescence mark. Acta Ecol. Sin. 2007, 27, 2468–2476. [Google Scholar]
- Qin, Y.j.; Krosch, M.N.; Schutze, M.K.; Zhang, Y.; Wang, X.X.; Prabhakar, C.S.; Susanto, A.; Hee, A.K.; Ekesi, S.; Badji, K. Population structure of a global agricultural invasive pest, Bactrocera dorsalis (Diptera: Tephritidae). Evol. Appl. 2018, 11, 1990–2003. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, J.S.; Naaz, N.; Prabhakar, C.S.; Lemtur, M. Genetic analysis of oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae) populations based on mitochondrial cox1 and nad1 gene sequences from India and other Asian countries. Genetica 2016, 144, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Allwood, A.; Chinajariyawong, A.; Kritsaneepaiboon, S.; Drew, R.; Hamacek, E.; Hancock, D.; Hengsawad, C.; Jipanin, J.; Jirasurat, M.; Krong, C.K. Host plant records for fruit flies (Diptera: Tephritidae) in Southeast Asia. Raffles Bull. Zool. 1999, 47, 1–92. [Google Scholar]
- Ekesi, S.; de Meyer, M.; Mohamed, S.A.; Virgilio, M.; Borgemeister, C. Taxonomy, ecology, and management of native and exotic fruit fly species in Africa. Annu. Rev. Entomol. 2016, 61, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Koyama, J.; Teruya, T.; Tanaka, K. Eradication of the oriental fruit fly (Diptera: Tephritidae) from the Okinawa Islands by a male annihilation method. J. Econ. Entomol. 1984, 77, 468–472. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, N.; Ji, Q.; Yang, J.; Zheng, M. Genetic structure analysis of three kinds of fruit fly of economic important in China. In Proceedings of the Ninth International Symposium on Fruit Flies of Economic Importance (9th ISFFEI), Bangkok, Thailand, 12–16 May 2014; pp. 12–16. [Google Scholar]
- Iwaizumi, R. Species and host record of the Bactrocera dorsalis complex (Diptera: Tephritidae) detected by the plant quarantine of Japan. Appl. Entomol. Zool. 2004, 39, 327–333. [Google Scholar] [CrossRef]
- Dai, S.; Lin, C.; Chang, C. Polymorphic microsatellite DNA markers from the oriental fruit fly Bactrocera dorsalis (Hendel). Mol. Ecol. Notes 2004, 4, 629–631. [Google Scholar] [CrossRef]
- Ohno, S.; Tamura, Y.; Haraguchi, D.; Matsuyama, T.; Kohama, T. Re-invasions by Bactrocera dorsalis complex (Diptera: Tephritidae) occurred after its eradication in Okinawa, Japan, and local differences found in the frequency and temporal patterns of invasions. Appl. Entomol. Zool. 2009, 44, 643–654. [Google Scholar] [CrossRef] [Green Version]
- KATI. Agricultural Products Export Information, Statistics of Import and Export. 2016. Available online: https://www.kati.net/index.do (accessed on 10 September 2021).
- Aketarawong, N.; Isasawin, S.; Sojikul, P.; Thanaphum, S. Gene flow and genetic structure of Bactrocera carambolae (Diptera, tephritidae) among geographical differences and sister species, B. dorsalis, inferred from microsatellite DNA data. ZooKeys 2015, 2015, 239–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, G.-F.; Kerrest, A.; Dujon, B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 2008, 72, 686–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.A.; Bravo, J.P.; Nobile, P.M.; Morelli, K.A. Microsatellites as tools for genetic diversity analysis. In Genetic Diversity in Microorganisms; Citeseer: Princeton, NY, USA, 2012. [Google Scholar]
- Burgess, T.I.; Sakalidis, M.L.; Hardy, G.E.S.J. Gene flow of the canker pathogen Botryosphaeria australis between Eucalyptus globulus plantations and native eucalypt forests in Western Australia. Austral Ecol. 2006, 31, 559–566. [Google Scholar] [CrossRef]
- Bonizzoni, M.; Zheng, L.; Guglielmino, C.; Haymer, D.; Gasperi, G.; Gomulski, L.; Malacrida, A. Microsatellite analysis of medfly bioinfestations in California. Mol. Ecol. 2001, 10, 2515–2524. [Google Scholar] [CrossRef]
- Bonizzoni, M.; Guglielmino, C.; Smallridge, C.; Gomulski, M.; Malacrida, A.; Gasperi, G. On the origins of medfly invasion and expansion in Australia. Mol. Ecol. 2004, 13, 3845–3855. [Google Scholar] [CrossRef]
- Meixner, M.D.; McPheron, B.A.; Silva, J.G.; Gasparich, G.E.; Sheppard, W.S. The Mediterranean fruit fly in California: Evidence for multiple introductions and persistent populations based on microsatellite and mitochondrial DNA variability. Mol. Ecol. 2002, 11, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Kerdelhué, C.; Ye, H. Genetic structure and inferences on potential source areas for Bactrocera dorsalis (Hendel) based on mitochondrial and microsatellite markers. PLoS ONE 2012, 7, e37083. [Google Scholar] [CrossRef] [Green Version]
- Khamis, F.M.; Karam, N.; Ekesi, S.; de Meyer, M.; Bonomi, A.; Gomulski, L.M.; Scolari, F.; Gabrieli, P.; Siciliano, P.; Masiga, D.; et al. Uncovering the tracks of a recent and rapid invasion: The case of the fruit fly pest Bactrocera invadens (Diptera: Tephritidae) in Africa. Mol. Ecol. 2009, 18, 4798–4810. [Google Scholar] [CrossRef]
- Wan, X.; Nardi, F.; Zhang, B.; Liu, Y. The oriental fruit fly, Bactrocera dorsalis, in China: Origin and gradual inland range expansion associated with population growth. PLoS ONE 2011, 6, e25238. [Google Scholar] [CrossRef] [Green Version]
- Aketarawong, N.; Guglielmino, C.; Karam, N.; Falchetto, M.; Manni, M.; Scolari, F.; Gomulski, L.; Gasperi, G.; Malacrida, A. The oriental fruitfly Bactrocera dorsalis s.s. in East Asia: Disentangling the different forces promoting the invasion and shaping the genetic make-up of populations. Genetica 2014, 142, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.; Ruiz-Arce, R.; McPheron, B.A.; Wu, J.; Li, Z. Microsatellite markers reveal population structure and low gene flow among collections of Bactrocera cucurbitae (Diptera: Tephritidae) in Asia. J. Econ. Entomol. 2011, 104, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, M.; Delatte, H.; Backeljau, T.; de Meyer, M. Macrogeographic population structuring in the cosmopolitan agricultural pest Bactrocera cucurbitae (Diptera: Tephritidae). Mol. Ecol. 2010, 19, 2713–2724. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Carapelli, A.; Dallai, R.; Roderick, G.K.; Frati, F. Population structure and colonization history of the olive fly, Bactrocera oleae (Diptera, Tephritidae). Mol. Ecol. 2005, 14, 2729–2738. [Google Scholar] [CrossRef]
- Dogaç, E.; Kandemir, İ.; Taskin, V. The genetic polymorphisms and colonization process of olive fly populations in Turkey. PLoS ONE 2013, 8, e56067. [Google Scholar] [CrossRef] [Green Version]
- Zygouridis, N.; Augustinos, A.; Zalom, F.; Mathiopoulos, K. Analysis of olive fly invasion in California based on microsatellite markers. Heredity 2009, 102, 402–412. [Google Scholar] [CrossRef] [Green Version]
- Barbara, T.; Palma-silva, C.; Paggi, G.M.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef]
- Shearman, D.; Gilchrist, A.; Crisafulli, D.; Graham, G.; Lange, C.; Frommer, M. Microsatellite markers for the pest fruit fly, Bactrocera papayae (Diptera: Tephritidae) and other Bactrocera species. Mol. Ecol. Resour. 2006, 6, 4–7. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, H.; Raphael, K.; Gilchrist, A. Genetic delineation of sibling species of the pest fruit fly Bactocera (Diptera: Tephritidae) using microsatellites. Bull. Entomol. Res. 2003, 93, 351–360. [Google Scholar] [CrossRef]
- Kinnear, M.; Bariana, H.; Sved, J.; Frommer, M. Polymorphic microsatellite markers for population analysis of a tephritid pest species, Bactrocera tryoni. Mol. Ecol. 1998, 7, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Augustinos, A.A.; Stratikopoulos, E.E.; Drosopoulou, E.; Kakani, E.G.; Mavragani-Tsipidou, P.; Zacharopoulou, A.; Mathiopoulos, K.D. Isolation and characterization of microsatellite markers from the olive fly, Bactrocera oleae, and their cross-species amplification in the Tephritidae family. BMC Genom. 2008, 9, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamis, F.; Karam, N.; Guglielmino, C.; Ekesi, S.; Masiga, D.; de Meyer, M.; Kenya, E.U.; Malacrida, A. Isolation and characterization of microsatellite markers in the newly discovered invasive fruit fly pest in Africa, Bactrocera invadens (Diptera: Tephritidae). Mol. Ecol. Resour. 2008, 8, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Krosch, M.N.; Schutze, M.K.; Armstrong, K.F.; Boontop, Y.; Boykin, L.M.; Chapman, T.A.; Englezou, A.; Cameron, S.L.; Clarke, A.R. Piecing together an integrative taxonomic puzzle: Microsatellite, wing shape and aedeagus length analyses of Bactrocera dorsalis sl (Diptera: Tephritidae) find no evidence of multiple lineages in a proposed contact zone along the Thai/Malay Peninsula. Syst. Entomol. 2013, 38, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Schutze, M.; Jessup, A.; Clarke, A.R. Wing shape as a potential discriminator of morphologically similar pest taxa within the Bactrocera dorsalis species complex (Diptera: Tephritidae). Bull. Entomol. Res. 2012, 102, 103–111. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Arnaud-Haond, S.; Belkhir, K. GENCLONE: A computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol. Ecol. Notes 2007, 7, 15–17. [Google Scholar] [CrossRef]
- Dorken, M.E.; Eckert, C.G. Severely reduced sexual reproduction in northern populations of a clonal plant, Decodonverticillatus (Lythraceae). J. Ecol. 2001, 89, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Raymond, M. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Barton, N.; Slatkin, M. A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 1986, 56, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Chwee, C.P. Increasing Consumption of Tropical and Subtropical Fruits. 2004. Available online: http://www.fao.org/fileadmin/templates/agphome/documents/horticulture/WHO/seoul/ITF_Executive_Summary.pdf (accessed on 10 September 2021).
- Rice, W.R. Analyzing Tables of Statistical Tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, A Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3.2); Institute of Ecology, University of Lausanne: Lausanne, Switzerland, 2002; Available online: http//www.unil.ch/izea/softwares/fstat.html (accessed on 10 September 2021).
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Pearse, D.E.; Crandall, K.A. Beyond FST: Analysis of population genetic data for conservation. Conserv. Genet. 2004, 5, 585–602. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and Power Analysis of Two Tests for Detecting Recent Population Bottlenecks from Allele Frequency Data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; vonHoldt, B.M. Structure harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. J. Genet Genom. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Piry, S.; Alapetite, A.; Cornuet, J.-M.; Paetkau, D.; Baudouin, L.; Estoup, A. GENECLASS2: A software for genetic assignment and first-generation migrant detection. J. Hered. 2004, 95, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Mountain, J.L. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Langella, O. Populations 1.2.31. Population Genetic Software (Individuals or Populations Distances, Phylogenetic Trees); Free Software Foundation, Inc.: Boston, MA, USA, 1999. [Google Scholar]
- Cornuet, J.M.; Santos, F.; Beaumont, M.A.; Robert, C.P.; Marin, J.M.; Balding, D.J.; Guillemaud, T.; Estoup, A. Inferring population history with DIY ABC: A user-friendly approach to approximate Bayesian computation. Bioinformatics 2008, 24, 2713–2719. [Google Scholar] [CrossRef] [Green Version]
- Estoup, A.; Guillemaud, T. Reconstructing routes of invasion using genetic data: Why, how and so what? Mol. Ecol. 2010, 19, 4113–4130. [Google Scholar] [CrossRef]
- Cornuet, J.-M.; Ravigné, V.; Estoup, A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1. 0). BMC Bioinform. 2010, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, L.; Kabashima, J.N. Pest Notes: Red Imported Fire Ant; UC Statewide IPM Program, University of California: St. Davis, CA, USA, 2013; Volume 7487, pp. 1–4. [Google Scholar]
- Mau, R.; Jang, E.; Vargas, R. The Hawaii area-wide fruit fly pest management programme. In Area-Wide Control of Insect Pests; Springer: Berlin/Heidelberg, Germany, 2007; pp. 671–683. [Google Scholar]
- Zeng, Y.; Reddy, G.V.; Li, Z.; Qin, Y.; Wang, Y.; Pan, X.; Jiang, F.; Gao, F.; Zhao, Z.H. Global distribution and invasion pattern of oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). J. Appl. Entomol. 2019, 143, 165–176. [Google Scholar] [CrossRef]
- Weems, H.; Heppner, J.; Nation, J.; Fasulo, T. Oriental fruit fly, Bactrocera dorsalis (Hendel) (Insecta: Diptera: Tephritidae). Featured Creatures: Entomology and Nematology; IFAS University of Florida: Gainesville, FL, USA, 2012; Volume 21. [Google Scholar]
- Goergen, G.; Vayssières, J.-F.; Gnanvossou, D.; Tindo, M. Bactrocera invadens (Diptera: Tephritidae), a new invasive fruit fly pest for the Afrotropical region: Host plant range and distribution in West and Central Africa. Environ. Entomol. 2011, 40, 844–854. [Google Scholar] [CrossRef]
- Vargas, R.I.; Leblanc, L.; Putoa, R.; Eitam, A. Impact of introduction of Bactrocera dorsalis (Diptera: Tephritidae) and classical biological control releases of Fopius arisanus (Hymenoptera: Braconidae) on economically important fruit flies in French Polynesia. J. Econ. Entomol. 2007, 100, 670–679. [Google Scholar] [CrossRef]
- Schutze, M.K.; Krosch, M.N.; Armstrong, K.F.; Chapman, T.A.; Englezou, A.; Chomič, A.; Cameron, S.L.; Hailstones, D.; Clarke, A.R. Population structure of Bactrocera dorsalis ss, B. papayae and B. philippinensis (Diptera: Tephritidae) in southeast Asia: Evidence for a single species hypothesis using mitochondrial DNA and wing-shape data. BMC Evol. Biol. 2012, 12, 130. [Google Scholar] [CrossRef] [Green Version]
- Shelly, T.E.; Pahio, E.; Edu, J. Synergistic and inhibitory interactions between methyl eugenol and cue lure influence trap catch of male fruit flies, Bactrocera dorsalis (Hendel) and B. cucurbitae (Diptera: Tephritidae). Fla. Entomol. 2004, 87, 481–486. [Google Scholar] [CrossRef]
- Ovaskainen, O.; Sato, K.; Bascompte, J.; Hanski, I. Metapopulation models for extinction threshold in spatially correlated landscapes. J. Theor. Biol. 2002, 215, 95–108. [Google Scholar] [CrossRef]
- Provine, W.B. Ernst Mayr: Genetics and speciation. Genetics 2004, 167, 1041–1046. [Google Scholar] [CrossRef]
- Kiritani, K.; Yamamura, K. Exotic insects and their pathways for invasion. In Invasive Species: Vectors and Management Strategies; Island Press: Washington, DC, USA, 2003; pp. 44–67. [Google Scholar]
- Marler, T.E. Increased threat of island endemic tree’s extirpation via invasion-induced decline of intrinsic resistance to recurring tropical cyclones. Commun. Integr. Biol. 2013, 6, e22361. [Google Scholar] [CrossRef]
- Landsea, C. What Regions Around the Globe Have Tropical Cyclones and Who is Responsible for Forecasting There; NOAA: Washington, DC, USA, 2014. [Google Scholar]
- Belles, J. Typhoon Alley: Where the Planet’s Most Intense Tropical Cyclones Most Frequently Happen; The Weather Company: Atlanta, GA, USA, 2016. [Google Scholar]
- Center, J.T.W. Tropical Cyclone Best Track Data Site. Available online: http://www.npmoc.navy.mil/jtwc/best_tracks 2006 (accessed on 10 September 2021).
- Otuka, A.; Nagayoshi, K.; Sanada-Morimura, S.; Matsumura, M.; Haraguchi, D.; Kakazu, R. Estimation of possible sources for wind-borne re-invasion of Bactrocera dorsalis complex (Diptera: Tephritidae) into islands of Okinawa Prefecture, southwestern Japan. Appl. Entomol. Zool. 2016, 51, 21–35. [Google Scholar] [CrossRef]
- Shoji, Y. Route by which non-living butterflies visit Japan. Butterflies 1995, 11, 48–54. [Google Scholar]
- Shi, W.; Kerdelhué, C.; Ye, H. Population genetic structure of the oriental fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae) from Yunnan province (China) and nearby sites across the border. Genetica 2010, 138, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Aketarawong, N.; Bonizzoni, M.; Malacrida, A.; Gasperi, G.; Thanaphum, S. Seventeen novel microsatellite markers from an enriched library of the pest species Bactrocera dorsalis sensu stricto. Mol. Ecol. Notes 2006, 6, 1138–1140. [Google Scholar] [CrossRef]
- Clarke, A.R.; Li, Z.h.; Qin, Y.j.; Zhao, Z.H.; Liu, L.j.; Schutze, M.K. Bactrocera dorsalis (Hendel) (Diptera: Tephritidae) is not invasive through Asia: It’s been there all along. J. Appl. Entomol. 2019, 143, 797–801. [Google Scholar] [CrossRef]
- Mohamed, Z.; AbdLatif, I.; Abdullah, A.M. Economic importance of tropical and subtropical fruits. In Postharvest Biology and Technology of Tropical and Subtropical Fruits; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1–20. [Google Scholar]
- Siddiq, M.; Ahmed, J.; Lobo, M.G.; Ozadali, F. Tropical and Subtropical Fruits: Postharvest Physiology, Processing and Packaging; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Yahia, E.M. Postharvest Biology and Technology of Tropical and Subtropical Fruits: Fundamental Issues; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Kader, A.; Yahia, E. Postharvest biology of tropical and subtropical fruits. In Postharvest Biology and Technology of Tropical and Subtropical Fruits; Elsevier: Amsterdam, The Netherlands, 2011; pp. 79–111. [Google Scholar]
- Melo-Ferreira, J.; Boursot, P.; Suchentrunk, F.; Ferrand, N.; Alves, P. Invasion from the cold past: Extensive introgression of mountain hare (Lepus timidus) mitochondrial DNA into three other hare species in northern Iberia. Mol. Ecol. 2005, 14, 2459–2464. [Google Scholar] [CrossRef]
- McGuire, J.A.; Linkem, C.W.; Koo, M.S.; Hutchison, D.W.; Lappin, A.K.; Orange, D.I.; Lemos-Espinal, J.; Riddle, B.R.; Jaeger, J.R. Mitochondrial introgression and incomplete lineage sorting through space and time: Phylogenetics of crotaphytid lizards. Evolution 2007, 61, 2879–2897. [Google Scholar] [CrossRef]
- Mastrantonio, V.; Porretta, D.; Urbanelli, S.; Crasta, G.; Nascetti, G. Dynamics of mtDNA introgression during species range expansion: Insights from an experimental longitudinal study. Sci. Rep. 2016, 6, 30355. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population ID | No. | GD | Na | Ne | Ho (± SD) | He (± SD) | HWE * | Hs | Rs | Fis |
|---|---|---|---|---|---|---|---|---|---|---|
| KR 001 | 9 | 1 | 5.27 | 3.93 | 0.681 (0.313) | 0.672 (0.216) | ns | 0.71 | 2.73 | 0.05 |
| KR 002 | 10 | 1 | 4.60 | 3.38 | 0.704 (0.240) | 0.636 (0.161) | ns | 0.67 | 2.55 | −0.05 |
| KR 003 | 5 | 1 | 5.07 | 4.16 | 0.667 (0.247) | 0.691 (0.216) | ns | 0.78 | 2.95 | 0.15 |
| KR 004 | 2 | 1 | 1.67 | 1.62 | 0.500 (0.500) | 0.283 (0.277) | ns | 0.32 | 1.67 | −0.58 |
| TW CHU | 7 | 1 | 4.40 | 3.27 | 0.571 (0.310) | 0.589 (0.242) | ns | 0.63 | 2.49 | 0.11 |
| TW JIA | 8 | 1 | 6.87 | 4.98 | 0.600 (0.223) | 0.751 (0.130) | ns | 0.82 | 3.06 | 0.26 |
| TW KAO | 20 | 1 | 8.73 | 4.93 | 0.657 (0.234) | 0.695 (0.238) | ns | 0.71 | 2.79 | 0.08 |
| TW TIC | 20 | 1 | 10.07 | 4.66 | 0.653 (0.177) | 0.740 (0.144) | ns | 0.76 | 2.90 | 0.14 |
| TW TIN | 20 | 1 | 10.20 | 5.30 | 0.573 (0.173) | 0.731 (0.185) | ns | 0.75 | 2.90 | 0.24 |
| TW TIP | 15 | 1 | 8.07 | 4.37 | 0.693 (0.201) | 0.682 (0.194) | ns | 0.71 | 2.73 | 0.02 |
| CN FJS | 24 | 1 | 7.27 | 4.27 | 0.522 (0.204) | 0.677 (0.230) | ns | 0.69 | 2.68 | 0.25 |
| CN SHA | 1 | 1 | 1.33 | 1.33 | 0.400 (0.507) | 0.200 (0.254) | NA | NA | NA | NA |
| CN YUN | 24 | 1 | 7.13 | 3.66 | 0.536 (0.231) | 0.605 (0.238) | ns | 0.62 | 2.47 | 0.14 |
| VN BC1 | 18 | 1 | 8.40 | 4.63 | 0.400 (0.193) | 0.678 (0.250) | ns | 0.71 | 2.76 | 0.43 |
| VN BC2 | 13 | 1 | 6.73 | 3.62 | 0.378 (0.254) | 0.650 (0.176) | ns | 0.69 | 2.63 | 0.45 |
| VN HCM | 18 | 1 | 6.53 | 3.34 | 0.581 (0.267) | 0.649 (0.196) | ns | 0.67 | 2.57 | 0.13 |
| LA VI1 | 24 | 1 | 7.60 | 3.68 | 0.514 (0.212) | 0.648 (0.225) | ns | 0.67 | 2.60 | 0.23 |
| LA VI2 | 24 | 1 | 8.20 | 3.55 | 0.489 (0.220) | 0.632 (0.237) | ns | 0.65 | 2.56 | 0.25 |
| LA VI3 | 24 | 1 | 8.40 | 3.95 | 0.461 (0.193) | 0.661 (0.221) | ns | 0.68 | 2.64 | 0.32 |
| MM YAN | 25 | 1 | 8.67 | 4.44 | 0.518 (0.224) | 0.723 (0.136) | ns | 0.74 | 2.81 | 0.30 |
| TH BAN | 5 | 1 | 3.93 | 3.17 | 0.640 (0.275) | 0.639 (0.132) | ns | 0.72 | 2.65 | 0.11 |
| TH CHA | 4 | 1 | 3.87 | 3.27 | 0.700 (0.215) | 0.665 (0.109) | ns | 0.77 | 2.80 | 0.09 |
| TH DA1 | 20 | 1 | 7.87 | 4.49 | 0.632 (0.232) | 0.703 (0.194) | ns | 0.72 | 2.78 | 0.13 |
| TH DA2 | 23 | 1 | 6.87 | 4.07 | 0.624 (0.219) | 0.715 (0.117) | ns | 0.74 | 2.78 | 0.16 |
| KH PNP | 1 | 1 | 1.40 | 1.40 | 0.400 (0.507) | 0.200 (0.254) | NA | NA | NA | NA |
| IN DEL | 9 | 1 | 5.53 | 4.02 | 0.559 (0.228) | 0.702 (0.142) | ns | 0.76 | 2.81 | 0.26 |
| NP POK | 1 | 1 | 1.40 | 1.40 | 0.400 (0.507) | 0.200 (0.254) | NA | NA | NA | NA |
| PH BUS | 4 | 1 | 3.40 | 2.78 | 0.567 (0.312) | 0.619 (0.100) | ns | 0.74 | 2.62 | 0.23 |
| PH GUG | 24 | 1 | 8.20 | 3.35 | 0.559 (0.221) | 0.641 (0.147) | ns | 0.66 | 2.54 | 0.15 |
| PH GUZ | 8 | 1 | 5.60 | 3.79 | 0.536 (0.226) | 0.685 (0.151) | ns | 0.75 | 2.78 | 0.28 |
| PH LU1 | 6 | 1 | 3.73 | 2.78 | 0.511 (0.299) | 0.548 (0.249) | ns | 0.61 | 2.38 | 0.16 |
| PH LU2 | 13 | 1 | 6.87 | 4.48 | 0.557 (0.272) | 0.708 (0.155) | ns | 0.75 | 2.81 | 0.25 |
| PH LU3 | 28 | 1 | 10.00 | 4.60 | 0.580 (0.169) | 0.739 (0.136) | ns | 0.76 | 2.87 | 0.23 |
| PH MIN | 13 | 1 | 6.40 | 4.13 | 0.582 (0.188) | 0.686 (0.173) | ns | 0.72 | 2.74 | 0.19 |
| PH PA1 | 6 | 1 | 4.80 | 3.78 | 0.549 (0.302) | 0.676 (0.148) | ns | 0.76 | 2.79 | 0.28 |
| PH PA2 | 22 | 1 | 7.13 | 3.83 | 0.524 (0.238) | 0.677 (0.163) | ns | 0.70 | 2.66 | 0.25 |
| PH PA3 | 8 | 1 | 5.60 | 3.99 | 0.633 (0.234) | 0.671 (0.178) | ns | 0.72 | 2.74 | 0.12 |
| PH PA4 | 9 | 1 | 5.80 | 3.71 | 0.566 (0.209) | 0.691 (0.115) | ns | 0.74 | 2.77 | 0.24 |
| PH PA5 | 33 | 1 | 7.40 | 3.88 | 0.590 (0.190) | 0.701 (0.105) | ns | 0.71 | 2.68 | 0.17 |
| MY PEN | 24 | 1 | 7.07 | 3.41 | 0.481 (0.222) | 0.619 (0.217) | ns | 0.64 | 2.48 | 0.24 |
| Case | Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation | p |
|---|---|---|---|---|---|---|
| 1 | Korea vs. East Asian country | |||||
| Among groups | 1 | 19.31 | 0.03550 | 0.61 | <0.0001 | |
| Among populations within groups | 38 | 857.701 | 0.61627 | 10.61 | <0.0001 | |
| Within populations | 1090 | 5620.326 | 5.15626 | 88.78 | 0.2176 | |
| Total | 1129 | 6497.337 | 5.80803 | |||
| 2 | Based on the STRUCTURE K = 2 scenario | |||||
| Among groups | 1 | 143.959 | 0.27097 | 4.57 | <0.0001 | |
| Among populations within groups | 38 | 733.053 | 0.50821 | 8.56 | <0.0001 | |
| Within populations | 1090 | 5620.326 | 5.15626 | 86.87 | <0.0001 | |
| Total | 1129 | 6497.337 | 5.93544 | |||
| 3 | Based on the PCoA and K = 3 scenario in STRUCTURE analysis | |||||
| Among groups | 2 | 310.919 | 0.36108 | 6.13 | <0.0001 | |
| Among populations within groups | 37 | 566.092 | 0.37043 | 6.29 | <0.0001 | |
| Within populations | 1090 | 5620.326 | 5.15626 | 87.58 | <0.0001 | |
| Total | 1129 | 6497.337 | 5.88777 | |||
| 4 | Based on sampling year, 2015 vs. 2016 vs. 2017 | |||||
| Among groups | 2 | 188.855 | 0.19165 | 3.28 | <0.0001 | |
| Among populations within groups | 37 | 688.157 | 0.49202 | 8.43 | <0.0001 | |
| Within populations | 1090 | 5620.326 | 5.15626 | 88.29 | <0.0001 | |
| Total | 1129 | 6497.337 | 5.83993 | |||
| 5 | Based on the longitude 113° E, west side vs. east side | |||||
| Among groups | 1 | 78.265 | 0.09072 | 1.56 | <0.0001 | |
| Among populations within groups | 38 | 798.746 | 0.57314 | 9.85 | <0.0001 | |
| Within populations | 1090 | 5620.326 | 5.15626 | 88.59 | 0.0036 | |
| Total | 1129 | 6497.337 | 5.82012 | |||
| Logistic Regression Closest | Scenario 1 | Scenario 2 | Scenario 3 | Scenario 4 | Scenario 5 | Scenario 6 |
|---|---|---|---|---|---|---|
| 6000 | 0.0495 [0.0302, 0.0689] | 0.0558 [0.0351, 0.0765] | 0.2944 [0.2495, 0.3393] | 0.2907 [0.2464, 0.3350] | 0.1674 [0.1331, 0.2017] | 0.1422 [0.1094, 0.1750] |
| 12,000 | 0.0448 [0.0317, 0.0579] | 0.0495 [0.0361, 0.0628] | 0.2954 [0.2623, 0.3286] | 0.2976 [0.2644, 0.3307] | 0.1673 [0.1421, 0.1925] | 0.1454 [0.1211, 0.1698] |
| 18,000 | 0.0426 [0.0322, 0.0529] | 0.0468 [0.0363, 0.0573] | 0.2982 [0.2703, 0.3261] | 0.3031 [0.2750, 0.3312] | 0.1644 [0.1436, 0.1852] | 0.1450 [0.1248, 0.1652] |
| 24,000 | 0.0414 [0.0325, 0.0503] | 0.0469 [0.0377, 0.0561] | 0.2994 [0.2747, 0.3241] | 0.3059 [0.2809, 0.3308] | 0.1621 [0.1440, 0.1802] | 0.1443 [0.1266, 0.1620] |
| 30,000 | 0.0398 [0.0321, 0.0476] | 0.0472 [0.0389, 0.0555] | 0.3003 [0.2779, 0.3227] | 0.3076 [0.2848, 0.3304] | 0.1611 [0.1448, 0.1774] | 0.1439 [0.1280, 0.1599] |
| 36,000 | 0.0388 [0.0319, 0.0458] | 0.0473 [0.0397, 0.0549] | 0.3011 [0.2803, 0.3219] | 0.3093 [0.2882, 0.3305] | 0.1597 [0.1448, 0.1747] | 0.1437 [0.1290, 0.1584] |
| 42,000 | 0.0378 [0.0315, 0.0442] | 0.0474 [0.0404, 0.0545] | 0.3014 [0.2820, 0.3208] | 0.3111 [0.2913, 0.3309] | 0.1588 [0.1448, 0.1727] | 0.1434 [0.1297, 0.1572] |
| 48,000 | 0.0370 [0.0312, 0.0429] | 0.0477 [0.0411, 0.0543] | 0.3019 [0.2835, 0.3202] | 0.3119 [0.2931, 0.3306] | 0.1582 [0.1451, 0.1713] | 0.1433 [0.1304, 0.1563] |
| 54,000 | 0.0366 [0.0311, 0.0420] | 0.0477 [0.0414, 0.0539] | 0.3014 [0.2840, 0.3188] | 0.3125 [0.2947, 0.3303] | 0.1583 [0.1458, 0.1707] | 0.1436 [0.1313, 0.1559] |
| 60,000 | 0.0361 [0.0310, 0.0412] | 0.0476 [0.0416, 0.0535] | 0.3008 [0.2842, 0.3174] | 0.3135 [0.2965, 0.3306] | 0.1581 [0.1463, 0.1700] | 0.1439 [0.1321, 0.1556] |
| Analysis | Subgroup 1 | Subgroup 2 | Subgroup 3 | Subgroup 4 | Scenario 1 | Scenario 2 | Scenario 3 |
|---|---|---|---|---|---|---|---|
| B-1 | CN + VN | TW + TH | PH + MY + IN | KR 001 | 0.0050 [0.0026, 0.0074] | 0.0015 [0.0007, 0.0023] | 0.9935 [0.9907, 0.9964] |
| B-2 | CN + VN | TW + TH | PH + MY + IN | KR 002 | 0.0040 [0.0026, 0.0053] | 0.0023 [0.0014, 0.0032] | 0.9937 [0.9919, 0.9956] |
| B-3 | CN + VN | TW + TH | PH + MY + IN | KR 003 | 0.9552 [0.9458, 0.9646] | 0.0428 [0.0335, 0.0521] | 0.0020 [0.0011, 0.0029] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Kim, S.; Kim, S.; Lee, Y.; Lee, H.-S.; Lee, S.-J.; Choi, D.-S.; Jeon, J.; Lee, J.-H. Population Genetics for Inferring Introduction Sources of the Oriental Fruit Fly, Bactrocera dorsalis: A Test for Quarantine Use in Korea. Insects 2021, 12, 851. https://doi.org/10.3390/insects12100851
Kim H, Kim S, Kim S, Lee Y, Lee H-S, Lee S-J, Choi D-S, Jeon J, Lee J-H. Population Genetics for Inferring Introduction Sources of the Oriental Fruit Fly, Bactrocera dorsalis: A Test for Quarantine Use in Korea. Insects. 2021; 12(10):851. https://doi.org/10.3390/insects12100851
Chicago/Turabian StyleKim, Hyojoong, Sohee Kim, Sangjin Kim, Yerim Lee, Heung-Sik Lee, Seong-Jin Lee, Deuk-Soo Choi, Jaeyong Jeon, and Jong-Ho Lee. 2021. "Population Genetics for Inferring Introduction Sources of the Oriental Fruit Fly, Bactrocera dorsalis: A Test for Quarantine Use in Korea" Insects 12, no. 10: 851. https://doi.org/10.3390/insects12100851
APA StyleKim, H., Kim, S., Kim, S., Lee, Y., Lee, H.-S., Lee, S.-J., Choi, D.-S., Jeon, J., & Lee, J.-H. (2021). Population Genetics for Inferring Introduction Sources of the Oriental Fruit Fly, Bactrocera dorsalis: A Test for Quarantine Use in Korea. Insects, 12(10), 851. https://doi.org/10.3390/insects12100851