
Analysis of the Genetic Variation of the Fruitless Gene within the Anopheles gambiae (Diptera: Culicidae) Complex Populations in Africa
 , ,
, , 
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genomics Data and Mosquito Collection
2.2. Data Analysis
2.2.1. Genetic Variation Analysis
2.2.2. Conservation of the fru Gene
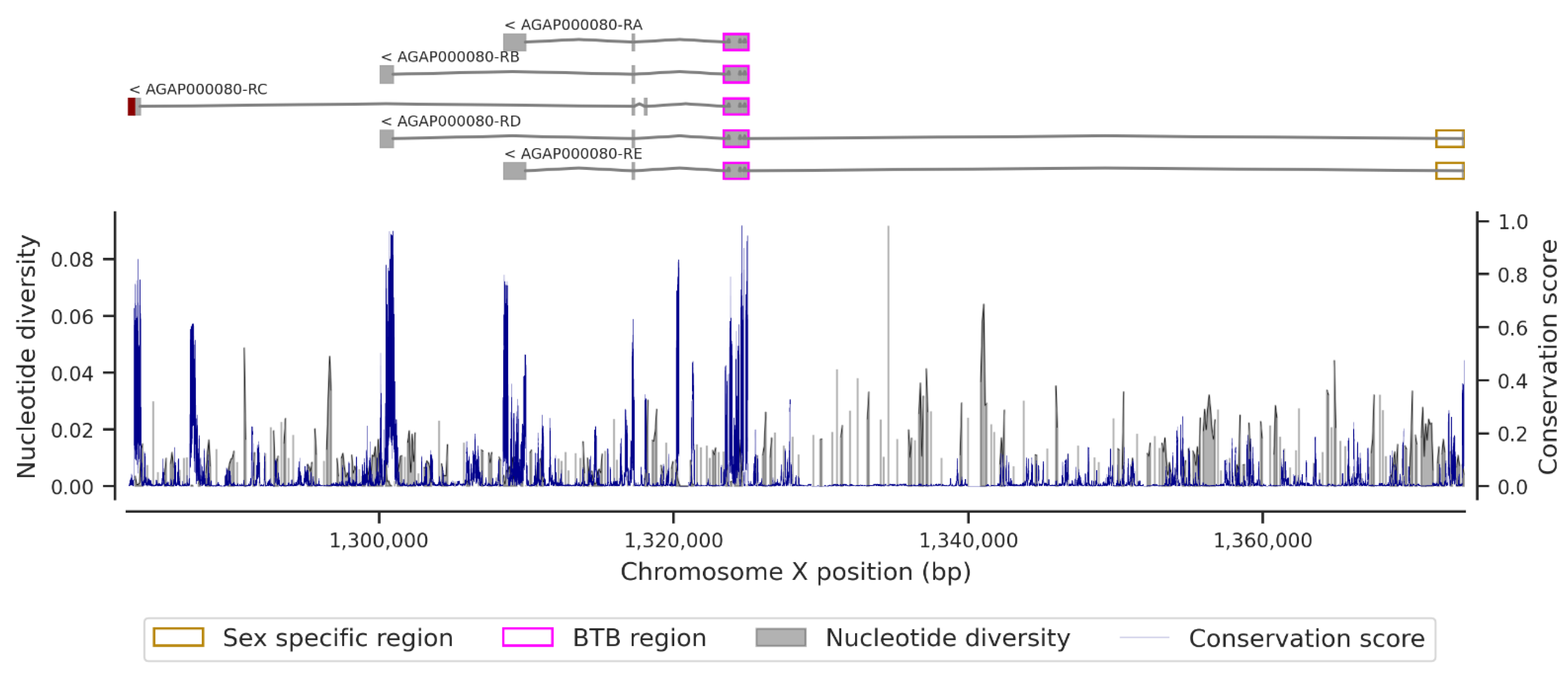
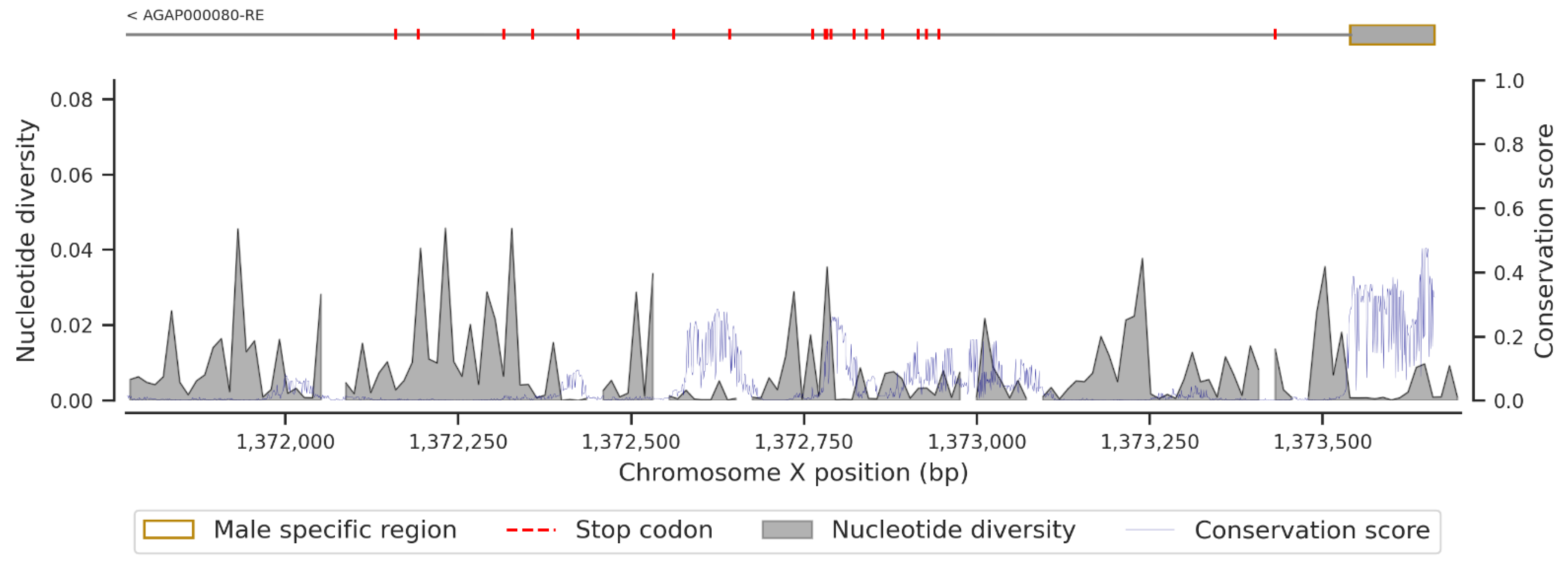
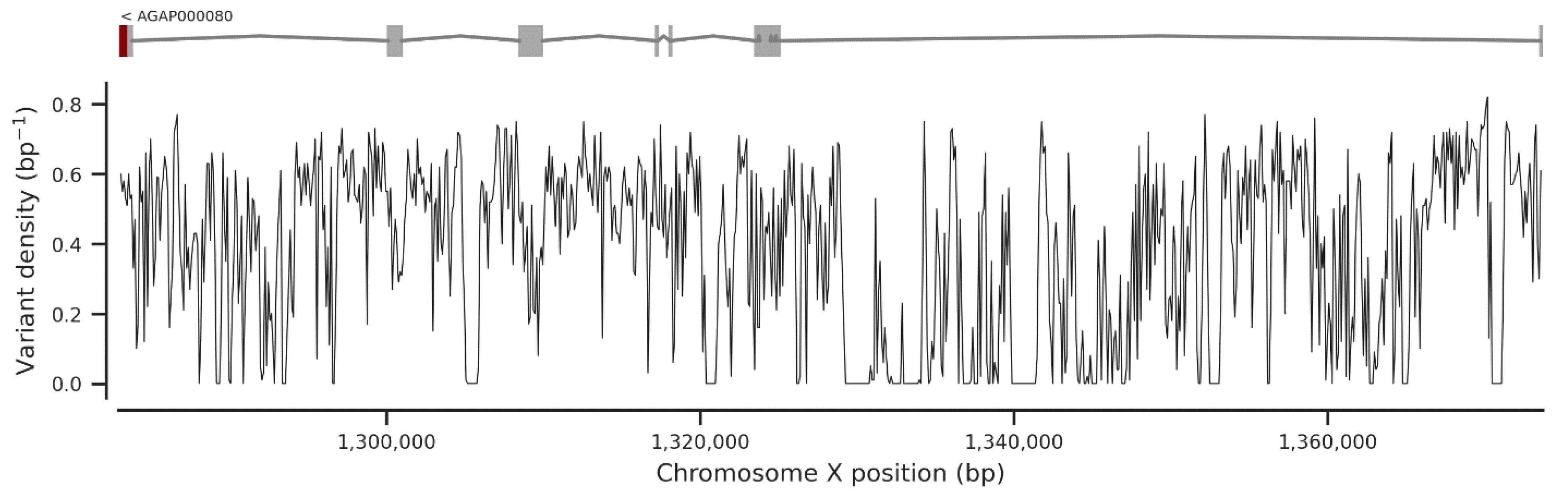
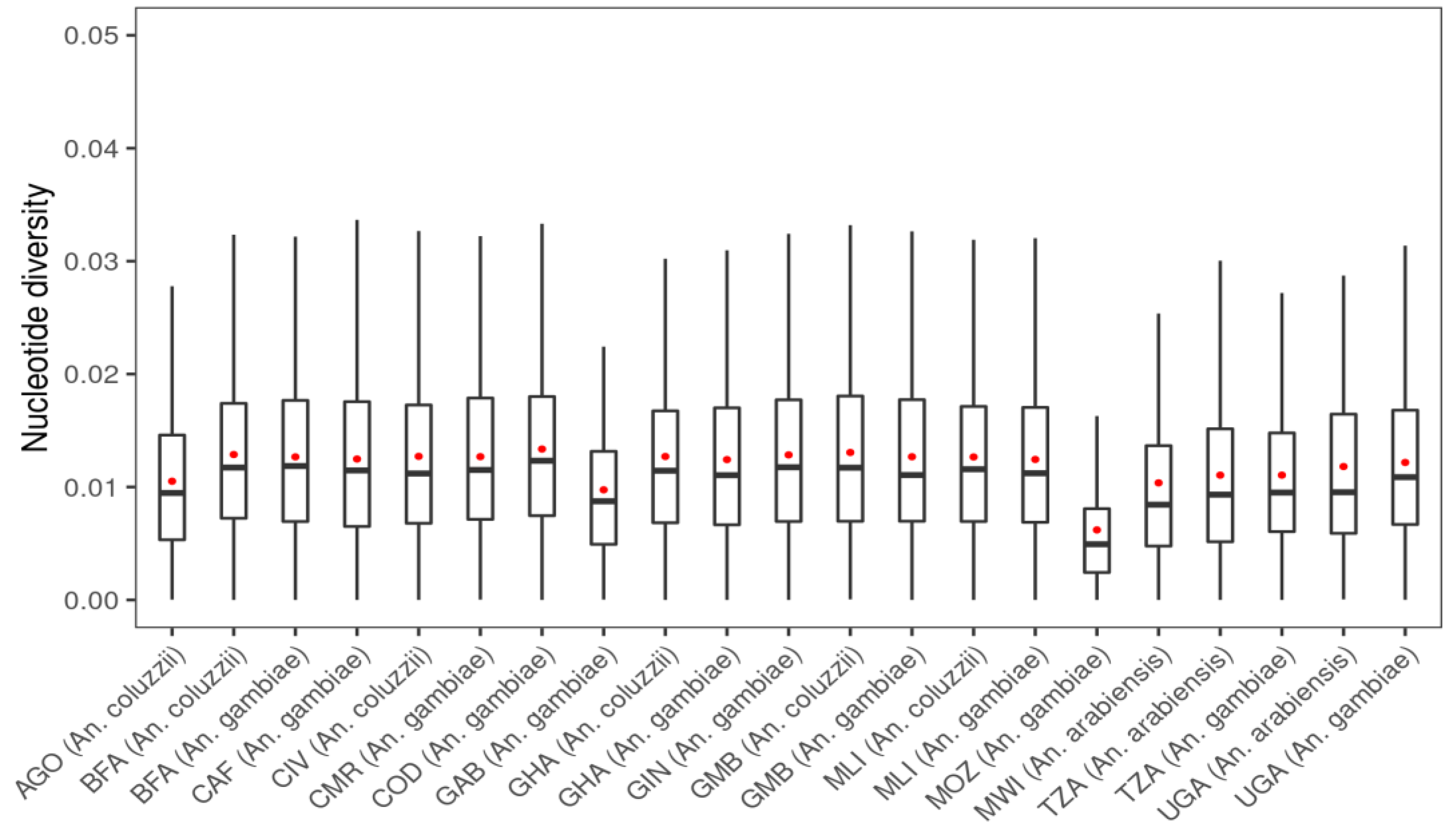
3. Results
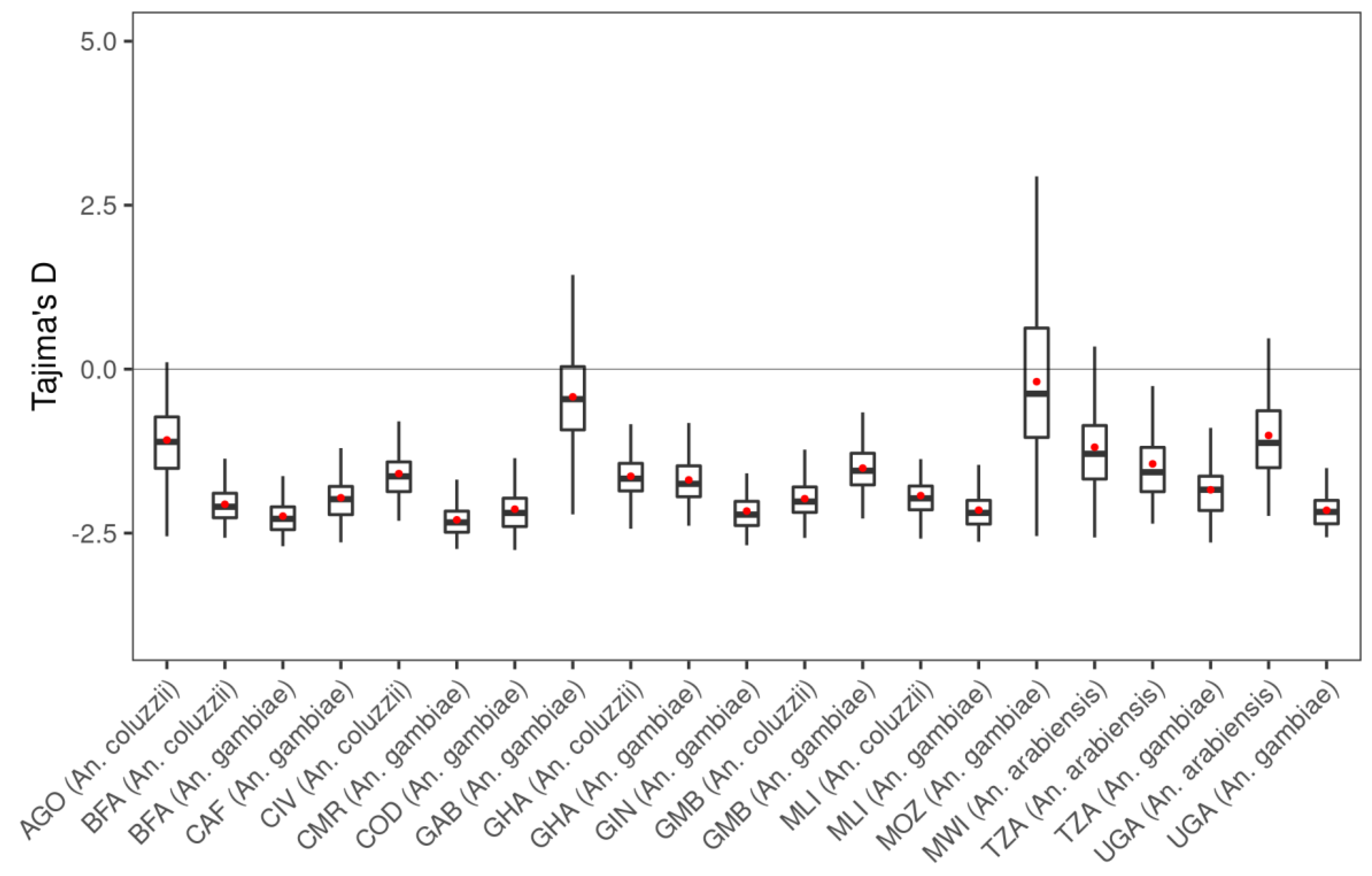
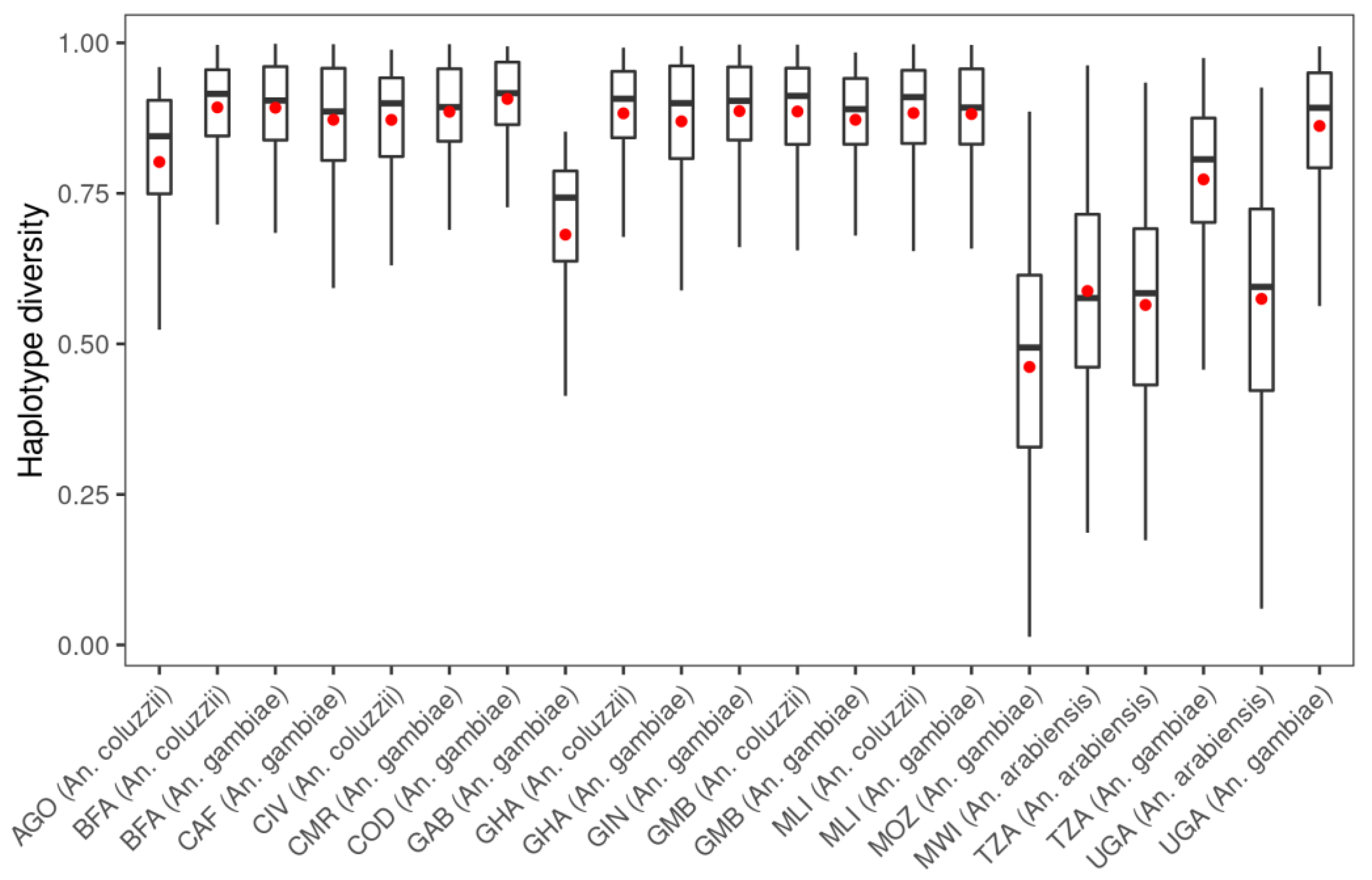
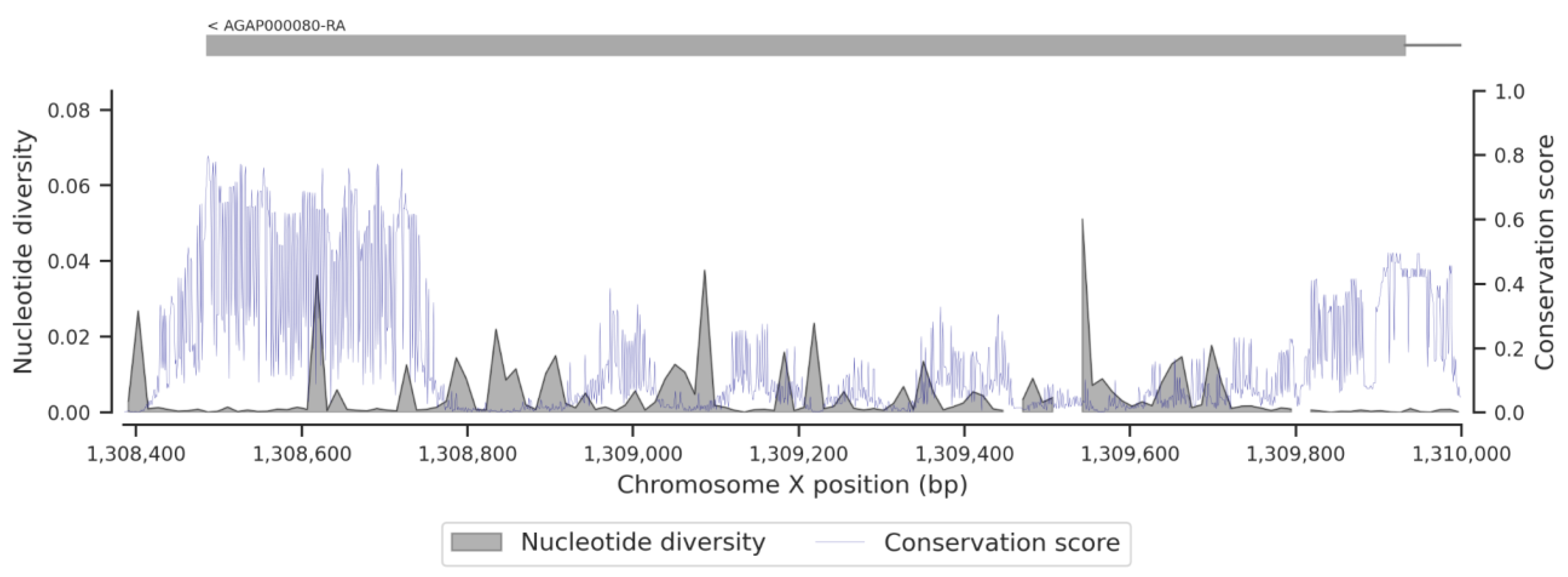
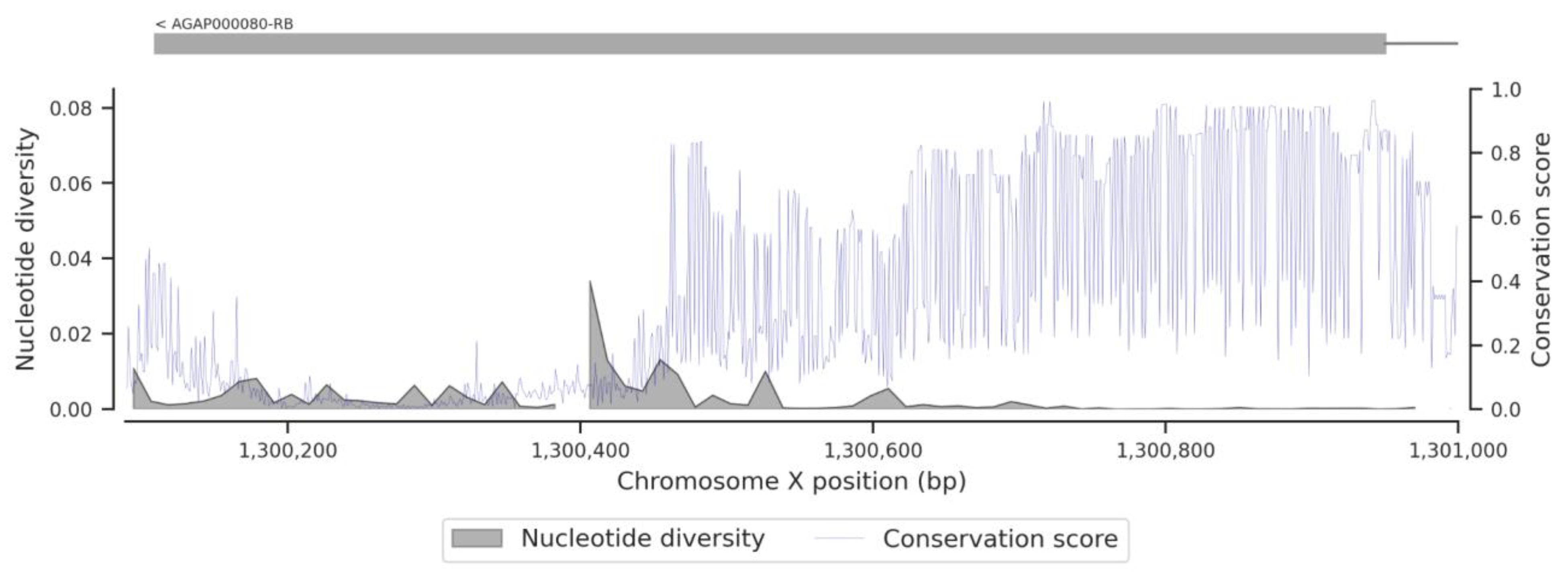
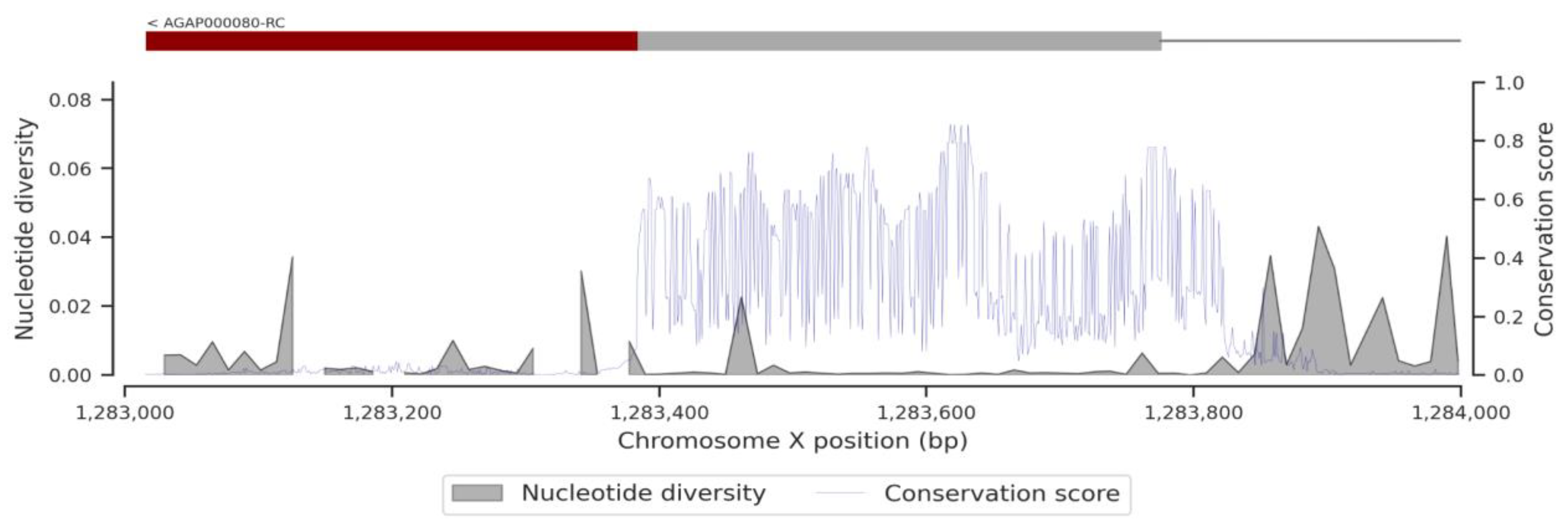
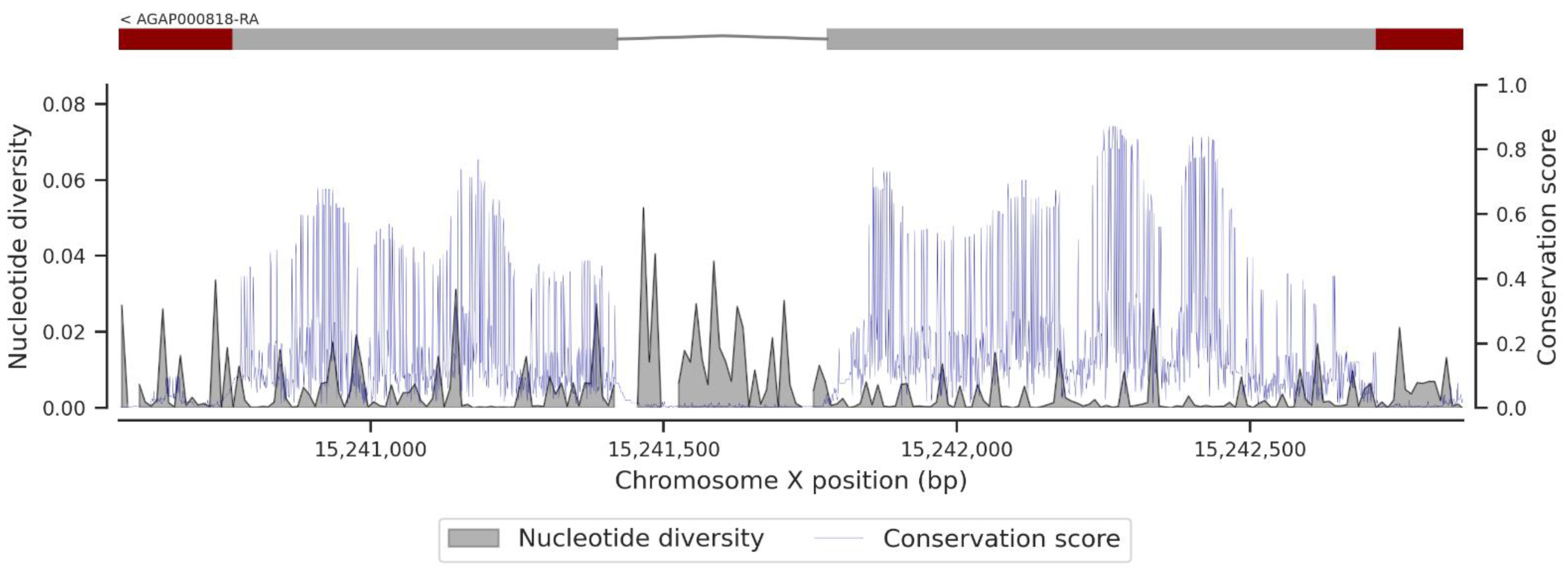
3.1. Genetic Variation within the fru Gene
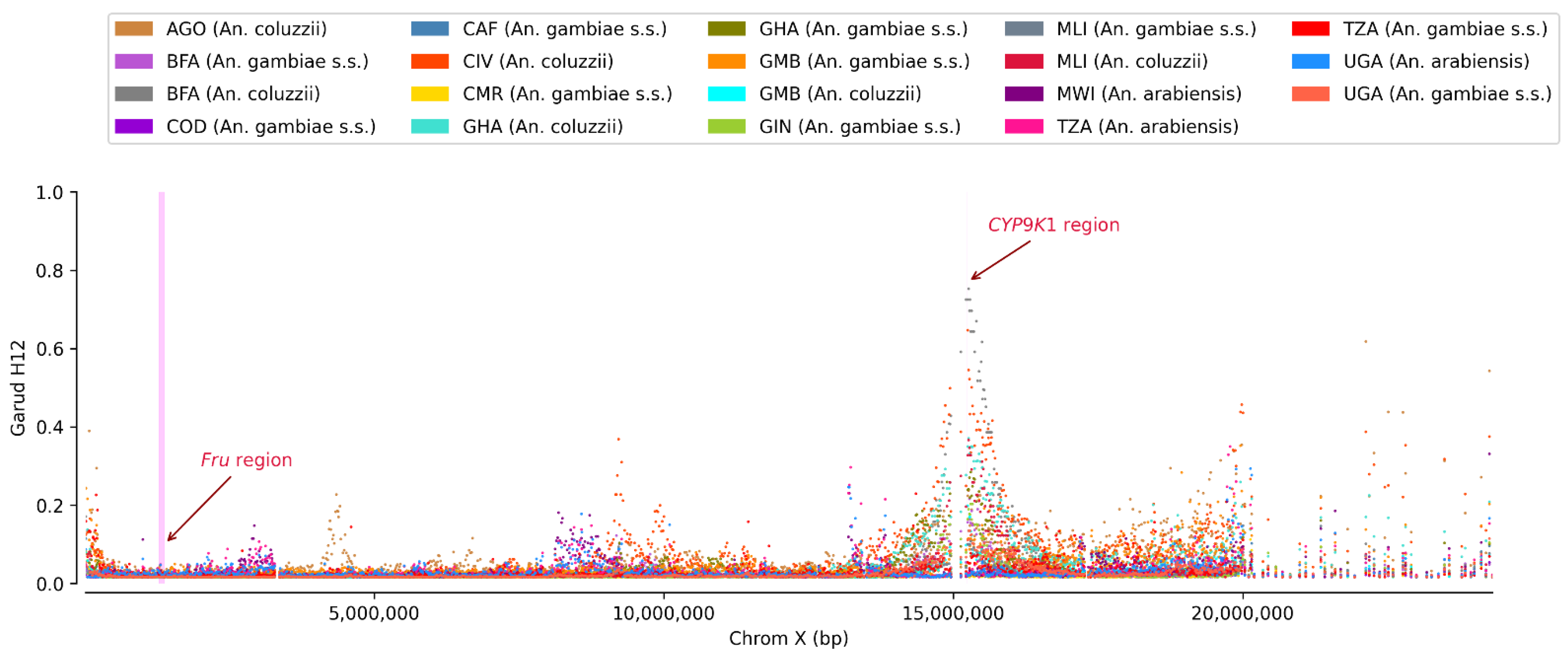
3.2. Non-Synonymous SNPs’ Variation
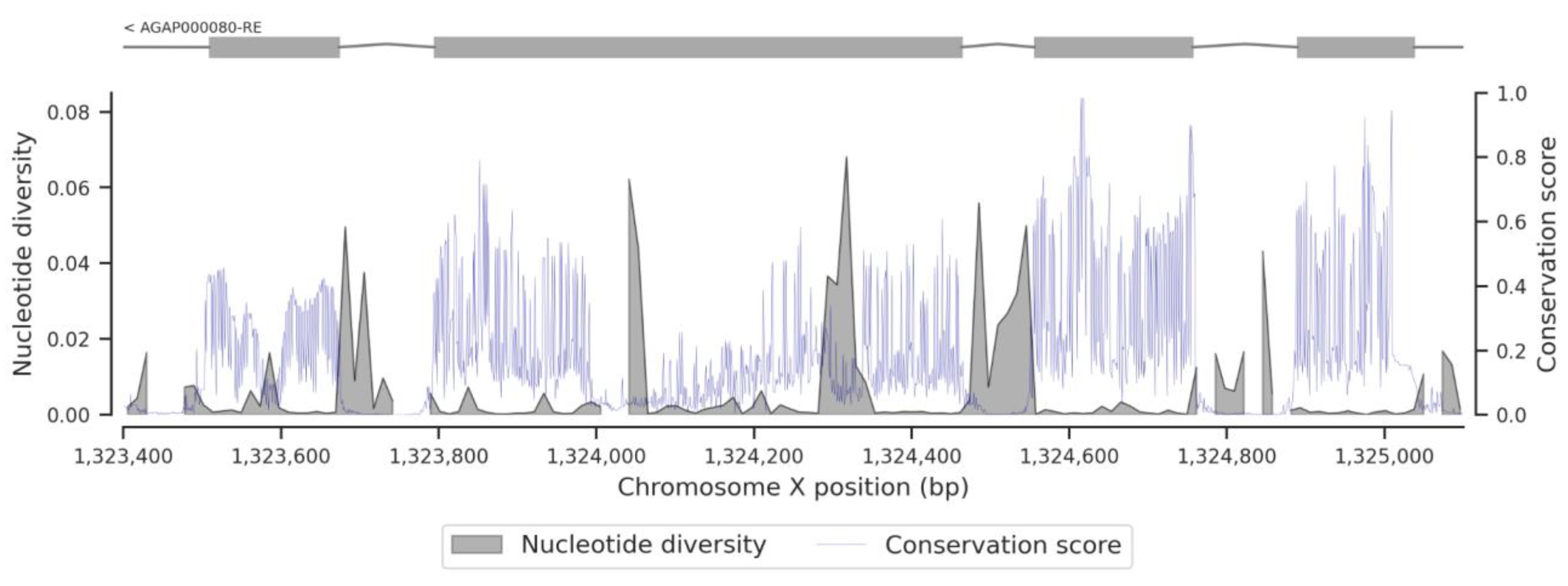
3.3. Conservation Score of the fru Gene
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A










References
- Sánchez, L. Sex-determining mechanisms in insects. Int. J. Dev. Biol. 2008, 52, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Siwicki, K.K.; Kravitz, E.A. Fruitless, Doublesex and the genetics of social behavior in Drosophila melanogaster. Curr. Opin. Neurobiol. 2009, 19, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, M.; Mauro, U.; Lombardo, F.; Milano, A.; Zazzaro, V.; Arcà, B.; Polito, L.C.; Saccone, G. Genomic organization and splicing evolution of the Doublesex gene, a Drosophila regulator of sexual differentiation, in the dengue and yellow fever mosquito Aedes aegypti. BMC Evol. Biol. 2011, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Biedler, J.K.; Tu, Z. Sex determination in mosquitoes. In Advances in Insect Physiology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 37–66. ISBN 0065-2806. [Google Scholar]
- Krzywinska, E.; Krzywinski, J. Effects of stable ectopic expression of the primary sex determination gene yob in the mosquito Anopheles gambiae. Parasites Vectors 2018, 11, 648. [Google Scholar] [CrossRef]
- Krzywinska, E.; Dennison, N.J.; Lycett, G.J.; Krzywinski, J. A maleness gene in the malaria mosquito Anopheles gambiae. Science 2016, 353, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Gailey, D.A.; Taylor, B.J.; Hall, J.C. Elements of the Fruitless locus regulate development of the muscle of lawrence, a male-specific structure in the abdomen of Drosophila melanogaster adults. Development 1991, 113, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, M.; D’Amato, R.; Petrella, V.; Aceto, S.; Nimmo, D.; Neira, M.; Alphey, L.; Polito, L.C.; Saccone, G. The orthologue of the fruitfly sex behaviour gene Fruitless in the mosquito Aedes aegypti: Evolution of genomic organisation and alternative splicing. PLoS ONE 2013, 8, e48554. [Google Scholar] [CrossRef]
- Gailey, D.A.; Billeter, J.-C.; Liu, J.H.; Bauzon, F.; Allendorfer, J.B.; Goodwin, S.F. Functional conservation of the Fruitless male sex-determination gene across 250 myr of insect evolution. Mol. Biol. Evol. 2005, 23, 633–643. [Google Scholar] [CrossRef]
- Yamamoto, D.; Kohatsu, S. What does the Fruitless gene tell us about nature vs. nurture in the sex life of Drosophila? Fly 2017, 11, 139–147. [Google Scholar] [CrossRef]
- Carnevale, P.; Robert, V. Les anophèles: Biologie, transmission du Plasmodium et lutte antivectorielle; IRD Éditions: Marseille, France, 2009. [Google Scholar] [CrossRef]
- Coetzee, M. Key to the females of afrotropical Anopheles mosquitoes (Diptera: Culicidae). Malar. J. 2020, 19, 70. [Google Scholar] [CrossRef]
- Bhatt, S.; Weiss, D.J.; Cameron, E.; Bisanzio, D.; Mappin, B.; Dalrymple, U.; Battle, K.E.; Moyes, C.L.; Henry, A.; Eckhoff, P.A.; et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature 2015, 526, 207. [Google Scholar] [CrossRef] [PubMed]
- Ranson, H. Current and future prospects for preventing malaria transmission via the use of insecticides. Cold Spring Harb. Perspect. Med. 2017, 7, a026823. [Google Scholar] [CrossRef] [PubMed]
- Benelli, G.; Beier, J.C. Current vector control challenges in the fight against malaria. Acta Trop. 2017, 174, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.; Hemingway, J.; Gleave, K.A.; Wiebe, A.; Gething, P.W.; Moyes, C.L. Developing global maps of insecticide resistance risk to improve vector control. Malar. J. 2017, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Sougoufara, S.; Doucouré, S.; Sembéne, P.M.B.; Harry, M.; Sokhna, C. Challenges for malaria vector control in sub-saharan africa: Resistance and behavioral adaptations in Anopheles populations. J. Vector Borne Dis. 2017, 54, 4–15. [Google Scholar] [PubMed]
- Bamou, R.; Mbakop, L.R.; Kopya, E.; Ndo, C.; Awono-Ambene, P.; Tchuinkam, T.; Rono, M.K.; Mwangangi, J.; Antonio-Nkondjio, C. Changes in malaria vector bionomics and transmission patterns in the equatorial forest region of Cameroon between 2000 and 2017. Parasites Vectors 2018, 11, 1–13. [Google Scholar] [CrossRef]
- Adelman, Z.N.; Tu, Z. Control of mosquito-borne infectious diseases: Sex and gene drive. Trends Parasitol. 2016, 32, 219–229. [Google Scholar] [CrossRef]
- Kyrou, K.; Hammond, A.M.; Galizi, R.; Kranjc, N.; Burt, A.; Beaghton, A.K.; Nolan, T.; Crisanti, A. A CRISPR–Cas9 gene drive targeting Doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol. 2018, 36, 1062–1066. [Google Scholar] [CrossRef]
- Hammond, A.; Galizi, R.; Kyrou, K.; Simoni, A.; Siniscalchi, C.; Katsanos, D.; Gribble, M.; Baker, D.; Marois, E.; Russell, S.; et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat. Biotechnol. 2016, 34, 78–83. [Google Scholar] [CrossRef]
- Hammond, A.; Pollegioni, P.; Persampieri, T.; North, A.; Minuz, R.; Trusso, A.; Bucci, A.; Kyrou, K.; Morianou, I.; Simoni, A.; et al. Gene-drive suppression of mosquito populations in large cages as a bridge between lab and field. Nat. Commun. 2021, 12, 4589. [Google Scholar] [CrossRef]
- Basrur, N.S.; de Obaldia, M.E.; Morita, T.; Herre, M.; von Heynitz, R.K.; Tsitohay, Y.N.; Vosshall, L.B. Fruitless mutant male mosquitoes gain attraction to human odor. Elife 2020, 9, e63982. [Google Scholar] [CrossRef] [PubMed]
- Hammond, A.; Karlsson, X.; Morianou, I.; Kyrou, K.; Beaghton, A.; Gribble, M.; Kranjc, N.; Galizi, R.; Burt, A.; Crisanti, A.; et al. Regulating the expression of gene drives is key to increasing their invasive potential and the mitigation of resistance. PLOS Genet. 2021, 17, e1009321. [Google Scholar] [CrossRef] [PubMed]
- The Anopheles gambiae 1000 Genomes Consortium. Ag1000G Phase 3 SNP Data Release. Available online: https://www.malariagen.net/data/ag1000g-phase3-snp (accessed on 29 April 2022).
- MalariaGEN. Malariagen Vector Data User Guide. Available online: https://malariagen.github.io/vector-data/landing-page.html (accessed on 30 April 2022).
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks—A publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; IOP Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar] [CrossRef]
- Miles, A.; Bot, P.; Murillo, R.; Ralph, P.; Harding, N.; Pisupati, R.; Rae, S.; Millar, T. Cggh/Scikit-Allel, Version 1.3.3; Zenodo 2021. Available online: https://zenodo.org/record/4759368#.Y3BX0-RBw2w (accessed on 5 October 2022).
- Miles, A. GitHub—Malariagen/Malariagen-Data-Python: A Python Package Providing Functions for Accessing and Analysing MalariaGEN Data. Available online: https://github.com/malariagen/malariagen-data-python (accessed on 6 May 2022).
- Rocklin, M. Dask: Parallel computation with blocked algorithms and task scheduling. In Proceedings of the 14th Python in Science Conference, Austin, TX, USA, 6–12 July 2015; pp. 126–132. [Google Scholar]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28–30 June 2010; pp. 56–61. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with numpy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; O’Kane, D.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Cole, J.B.; Warmenhoven, J.; et al. Mwaskom/Seaborn, Version 0.8.1; Zenodo 2017. Available online: https://zenodo.org/record/883859#.Y3BVeuRBw2w (accessed on 5 October 2022). [CrossRef]
- R Core Team. R: The R Project for Statistical Computing; Version 4.1.3; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Garud, N.R.; Messer, P.W.; Buzbas, E.O.; Petrov, D.A. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015, 11, e1005004. [Google Scholar] [CrossRef]
- Lewontin, R.C. The interaction of selection and linkage. I. general considerations; heterotic models. Genetics 1964, 49, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Kranjc, N.; Crisanti, A.; Nolan, T.; Bernardini, F. Anopheles gambiae genome conservation as a resource for rational gene drive target site selection. Insects 2021, 12, 97. [Google Scholar] [CrossRef]
- The Anopheles gambiae 1000 Genomes Consortium. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 2017, 552, 96–100. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Vontas, J.; Grigoraki, L.; Morgan, J.; Tsakireli, D.; Fuseini, G.; Segura, L.; de Carvalho, J.N.; Nguema, R.; Weetman, D.; Slotman, M.A.; et al. Rapid selection of a pyrethroid metabolic enzyme Cyp9k1 by operational malaria control activities. Proc. Natl. Acad. Sci. USA 2018, 115, 4619–4624. [Google Scholar] [CrossRef] [PubMed]
- Zollman, S.; Godt, D.; Privé, G.G.; Couderc, J.L.; Laski, F.A. The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc. Natl. Acad. Sci. USA 1994, 91, 10717–10721. [Google Scholar] [CrossRef] [PubMed]
- The Anopheles gambiae 1000 Genomes Consortium. Genome variation and population structure among 1142 mosquitoes of the African Malaria Vector Species Anopheles gambiae and Anopheles coluzzii. Genome Res. 2020, 30, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.A.; Subramanian, G.M.; Halpern, A.; Sutton, G.G.; Charlab, R.; Nusskern, D.R.; Wincker, P.; Clark, A.G.; Ribeiro, J.C.; Wides, R.; et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science 2002, 298, 129–149. [Google Scholar] [CrossRef]
- de Roos, A.D.G. Origins of introns based on the definition of exon modules and their conserved interfaces. Bioinformatics 2005, 21, 2–9. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Carmel, L.; Csuros, M.; Koonin, E. V Origin and evolution of spliceosomal introns. Biol. Direct 2012, 7, 11. [Google Scholar] [CrossRef]
- Grau-Bové, X.; Tomlinson, S.; O’Reilly, A.O.; Harding, N.J.; Miles, A.; Kwiatkowski, D.; Donnelly, M.J.; Weetman, D. Evolution of the insecticide target rdl in African Anopheles is driven by interspecific and interkaryotypic introgression. Mol. Biol. Evol. 2020, 37, 2900–2917. [Google Scholar] [CrossRef]
- Clarkson, C.S.; Miles, A.; Harding, N.J.; O’Reilly, A.O.; Weetman, D.; Kwiatkowski, D.; Donnelly, M.J. The genetic architecture of target-site resistance to pyrethroid insecticides in the African malaria vectors Anopheles gambiae and Anopheles coluzzii. Mol. Ecol. 2021, 30, 5303–5317. [Google Scholar] [CrossRef]
- Sætre, G.-P.; Ravinet, M. Evolutionary Genetics: Concepts, Analysis, and Practice; Oxford University Press: Oxford, UK, 2019; ISBN 9780198830917. [Google Scholar]
- Weetman, D.; Steen, K.; Rippon, E.J.; Mawejje, H.D.; Donnelly, M.J.; Wilding, C.S. Contemporary gene flow between wild An. gambiae s.s. and An. arabiensis. Parasites Vectors 2014, 7, 345. [Google Scholar] [CrossRef]
- Neafsey, D.E.; Lawniczak, M.K.N.; Park, D.J.; Redmond, S.N.; Coulibaly, M.B.; Traoré, S.F.; Sagnon, N.; Costantini, C.; Johnson, C.; Wiegand, R.C.; et al. SNP genotyping defines complex gene-flow boundaries among African malaria vector mosquitoes. Science 2010, 330, 514–517. [Google Scholar] [CrossRef]
- Champer, J.; Liu, J.; Oh, S.Y.; Reeves, R.; Luthra, A.; Oakes, N.; Clark, A.G.; Messer, P.W. Reducing resistance allele formation in CRISPR gene drive. Proc. Natl. Acad. Sci. USA 2018, 115, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Unckless, R.L.; Clark, A.G.; Messer, P.W. Evolution of resistance against CRISPR/Cas9 gene drive. Genetics 2017, 205, 827–841. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations | Number of Mosquitoes | SNPs | ns SNPs | Biallelic ns SNP | π | D | θ | H |
|---|---|---|---|---|---|---|---|---|
| AGO (An. coluzzii) | 81 | 2280 | 96 | 95 | 0.0095 | −1.109 | 0.014 | 0.845 |
| BFA (An. arabiensis) | 3 | 289 | 2 | 2 | 0.0088 | −0.256 | 0.008 | 0.600 |
| BFA (An. coluzzii) | 135 | 8312 | 118 | 114 | 0.0118 | −2.098 | 0.037 | 0.915 |
| BFA (An. gambiae s.s.) | 157 | 11,035 | 212 | 202 | 0.0119 | −2.282 | 0.046 | 0.904 |
| CAF (An. coluzzii) | 18 | 2372 | 16 | 16 | 0.0108 | −1.469 | 0.018 | 0.910 |
| CAF (An. gambiae s.s.) | 55 | 5303 | 38 | 38 | 0.0115 | −1.983 | 0.029 | 0.886 |
| CIV (An. coluzzii) | 80 | 4440 | 38 | 37 | 0.0112 | −1.635 | 0.023 | 0.900 |
| CMR (An. arabiensis) | 2 | 283 | 2 | 2 | 0.0157 | 0.180 | 0.015 | 0.833 |
| CMR (An. coluzzii) | 26 | 259 | 19 | 18 | 0.0104 | −1.407 | 0.017 | 0.897 |
| CMR (An. gambiae s.s.) | 416 | 17,291 | 430 | 392 | 0.0115 | −2.337 | 0.059 | 0.893 |
| COD (An. gambiae s.s.) | 76 | 7955 | 172 | 167 | 0.0123 | −2.192 | 0.039 | 0.916 |
| FRA (An. gambiae s.s.) | 23 | 966 | 56 | 56 | 0.0050 | −1.164 | 0.008 | 0.618 |
| GAB (An. gambiae s.s.) | 69 | 1348 | 20 | 19 | 0.0087 | −0.458 | 0.010 | 0.743 |
| GHA (An. coluzzii) | 64 | 4285 | 34 | 34 | 0.0114 | −1.670 | 0.024 | 0.907 |
| GHA (An. gambiae s.s.) | 36 | 3685 | 18 | 18 | 0.0110 | −1.749 | 0.024 | 0.900 |
| GIN (An. coluzzii) | 11 | 930 | 5 | 5 | 0.0095 | −0.258 | 0.009 | 0.835 |
| GIN (An. gambiae s.s.) | 123 | 9083 | 118 | 111 | 0.0118 | −2.218 | 0.040 | 0.904 |
| GMB (An. coluzzii) | 169 | 8241 | 92 | 91 | 0.0117 | −2.019 | 0.034 | 0.912 |
| GMB (An. gambiae s.s.) | 69 | 3952 | 37 | 35 | 0.0111 | −1.549 | 0.022 | 0.890 |
| GNB (An. gambiae s.s.) | 29 | 3375 | 32 | 32 | 0.0117 | −1.696 | 0.022 | 0.900 |
| KEN (An. gambiae s.s.) | 28 | 1221 | 44 | 44 | 0.0074 | −1.001 | 0.010 | 0.732 |
| MLI (An. arabiensis) | 2 | 188 | 0 | 0 | 0.0090 | 0.135 | 0.008 | 0.667 |
| MLI (An. coluzzii) | 91 | 6451 | 67 | 67 | 0.0116 | −1.970 | 0.032 | 0.910 |
| MLI (An. gambiae s.s.) | 131 | 8888 | 125 | 121 | 0.0112 | −2.191 | 0.041 | 0.893 |
| MOZ (An. gambiae s.s.) | 74 | 651 | 6 | 6 | 0.0049 | −0.375 | 0.005 | 0.494 |
| MWI (An. arabiensis) | 41 | 1381 | 15 | 15 | 0.0084 | −1.290 | 0.014 | 0.576 |
| TZA (An. arabiensis) | 225 | 2317 | 41 | 41 | 0.0093 | −1.571 | 0.019 | 0.584 |
| TZA (An. gambiae s.s.) | 68 | 4066 | 71 | 70 | 0.0095 | −1.838 | 0.025 | 0.807 |
| UGA (An. arabiensis) | 82 | 1545 | 26 | 26 | 0.0095 | −1.124 | 0.015 | 0.595 |
| UGA (An. gambiae s.s.) | 207 | 10,083 | 120 | 114 | 0.0109 | −2.177 | 0.042 | 0.892 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kientega, M.; Kranjc, N.; Traoré, N.; Kaboré, H.; Soma, D.D.; Morianou, I.; Namountougou, M.; Belem, A.M.G.; Diabaté, A. Analysis of the Genetic Variation of the Fruitless Gene within the Anopheles gambiae (Diptera: Culicidae) Complex Populations in Africa. Insects 2022, 13, 1048. https://doi.org/10.3390/insects13111048
Kientega M, Kranjc N, Traoré N, Kaboré H, Soma DD, Morianou I, Namountougou M, Belem AMG, Diabaté A. Analysis of the Genetic Variation of the Fruitless Gene within the Anopheles gambiae (Diptera: Culicidae) Complex Populations in Africa. Insects. 2022; 13(11):1048. https://doi.org/10.3390/insects13111048
Chicago/Turabian StyleKientega, Mahamadi, Nace Kranjc, Nouhoun Traoré, Honorine Kaboré, Dieudonné Diloma Soma, Ioanna Morianou, Moussa Namountougou, Adrien Marie Gaston Belem, and Abdoulaye Diabaté. 2022. "Analysis of the Genetic Variation of the Fruitless Gene within the Anopheles gambiae (Diptera: Culicidae) Complex Populations in Africa" Insects 13, no. 11: 1048. https://doi.org/10.3390/insects13111048
APA StyleKientega, M., Kranjc, N., Traoré, N., Kaboré, H., Soma, D. D., Morianou, I., Namountougou, M., Belem, A. M. G., & Diabaté, A. (2022). Analysis of the Genetic Variation of the Fruitless Gene within the Anopheles gambiae (Diptera: Culicidae) Complex Populations in Africa. Insects, 13(11), 1048. https://doi.org/10.3390/insects13111048