
Comparative Mitogenome Analyses of Subgenera and Species Groups in Epeorus (Ephemeroptera: Heptageniidae)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Identification and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Mitogenome Annotation and Analyses
2.4. Phylogenetic Analysis
3. Results and Discussion
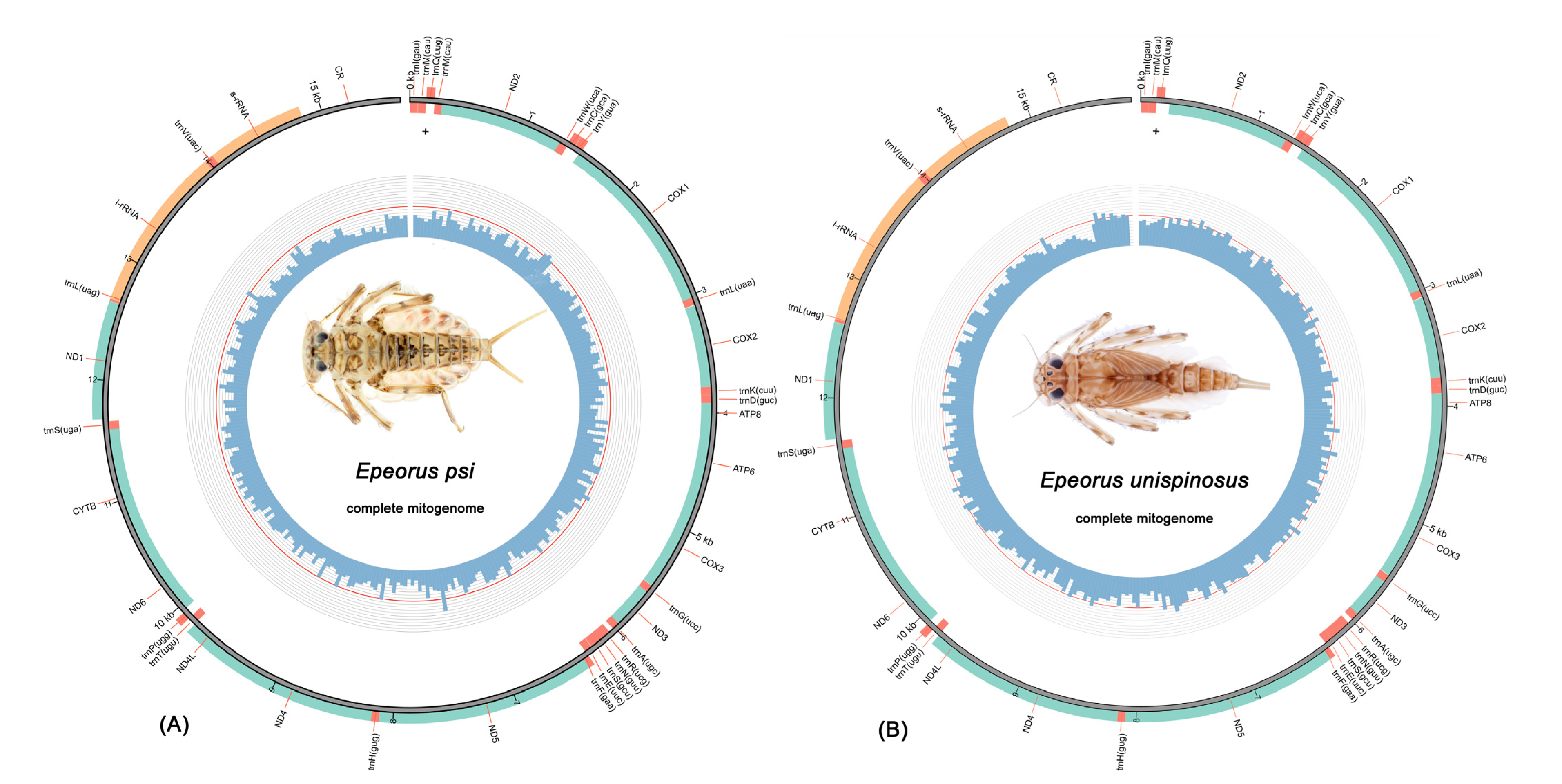
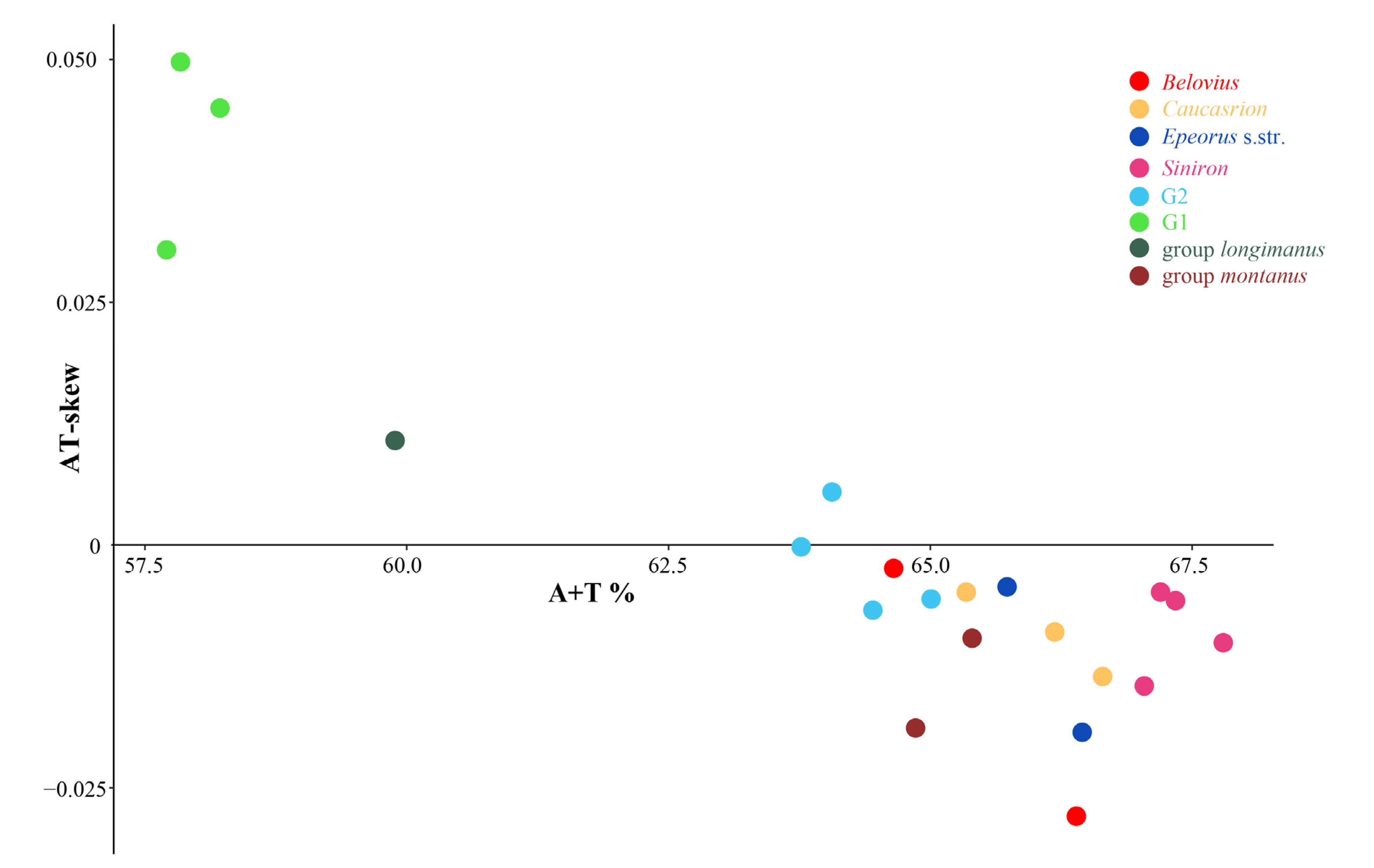
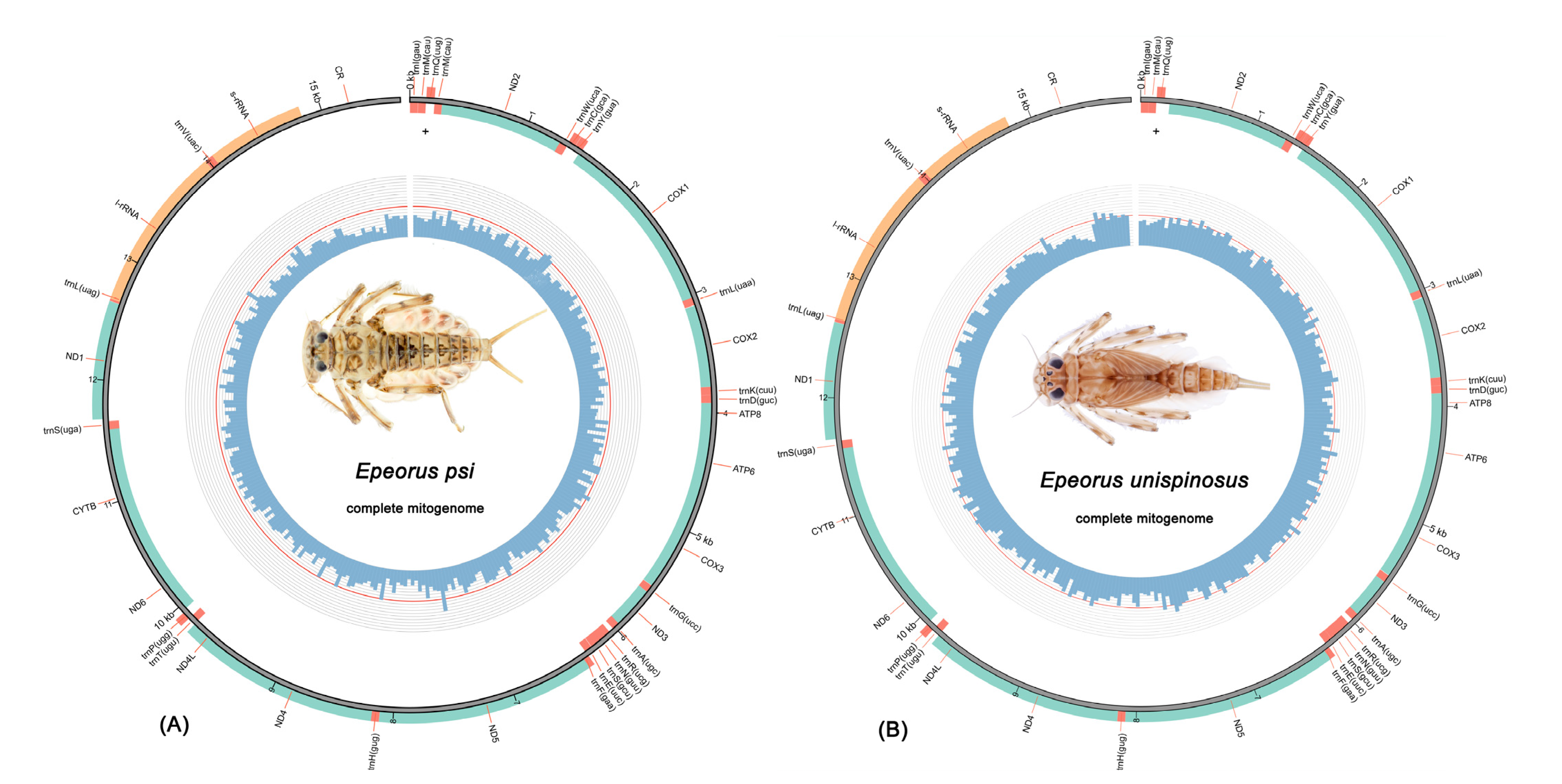
3.1. Mitogenome Organization and Composition
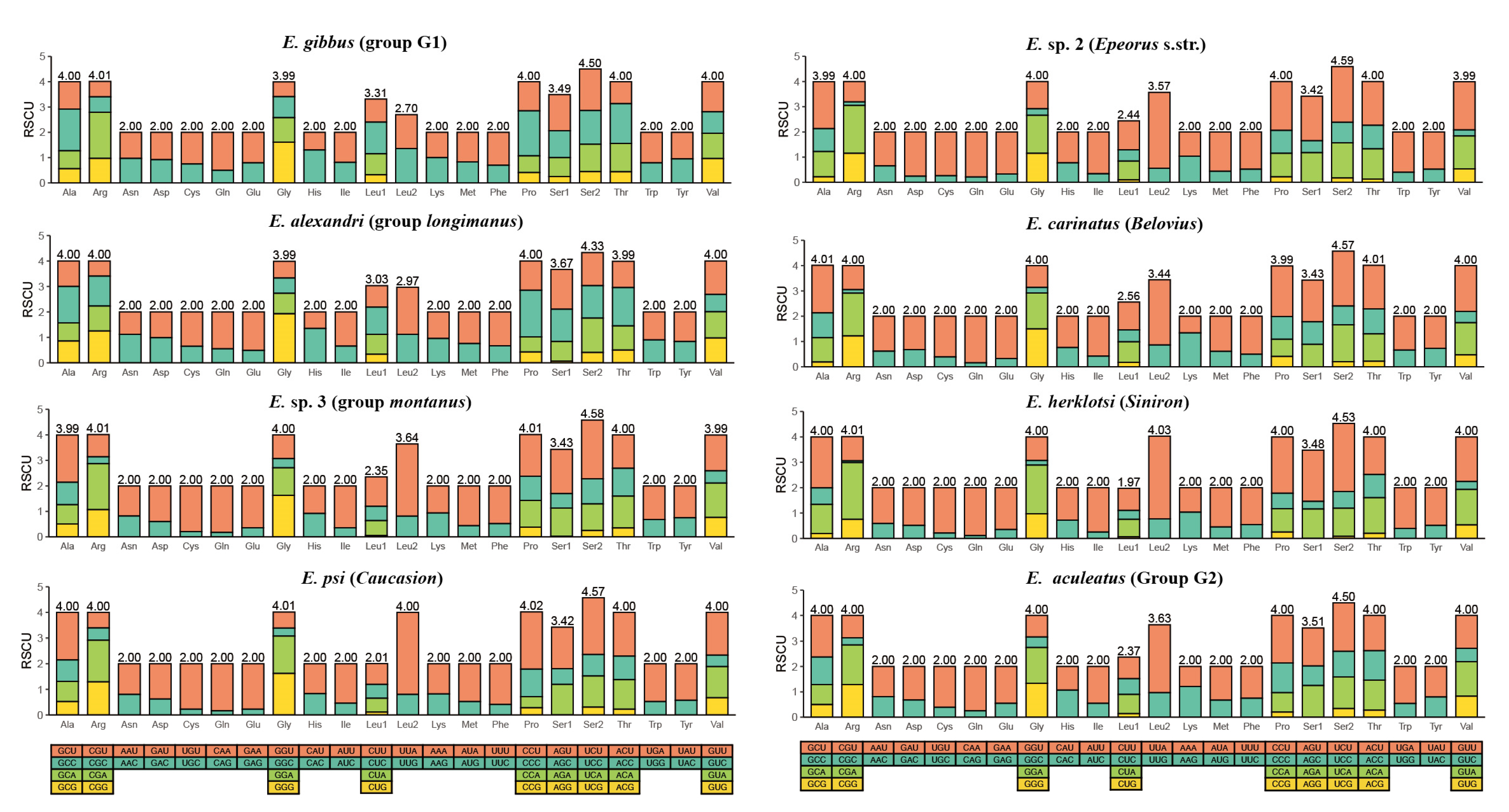
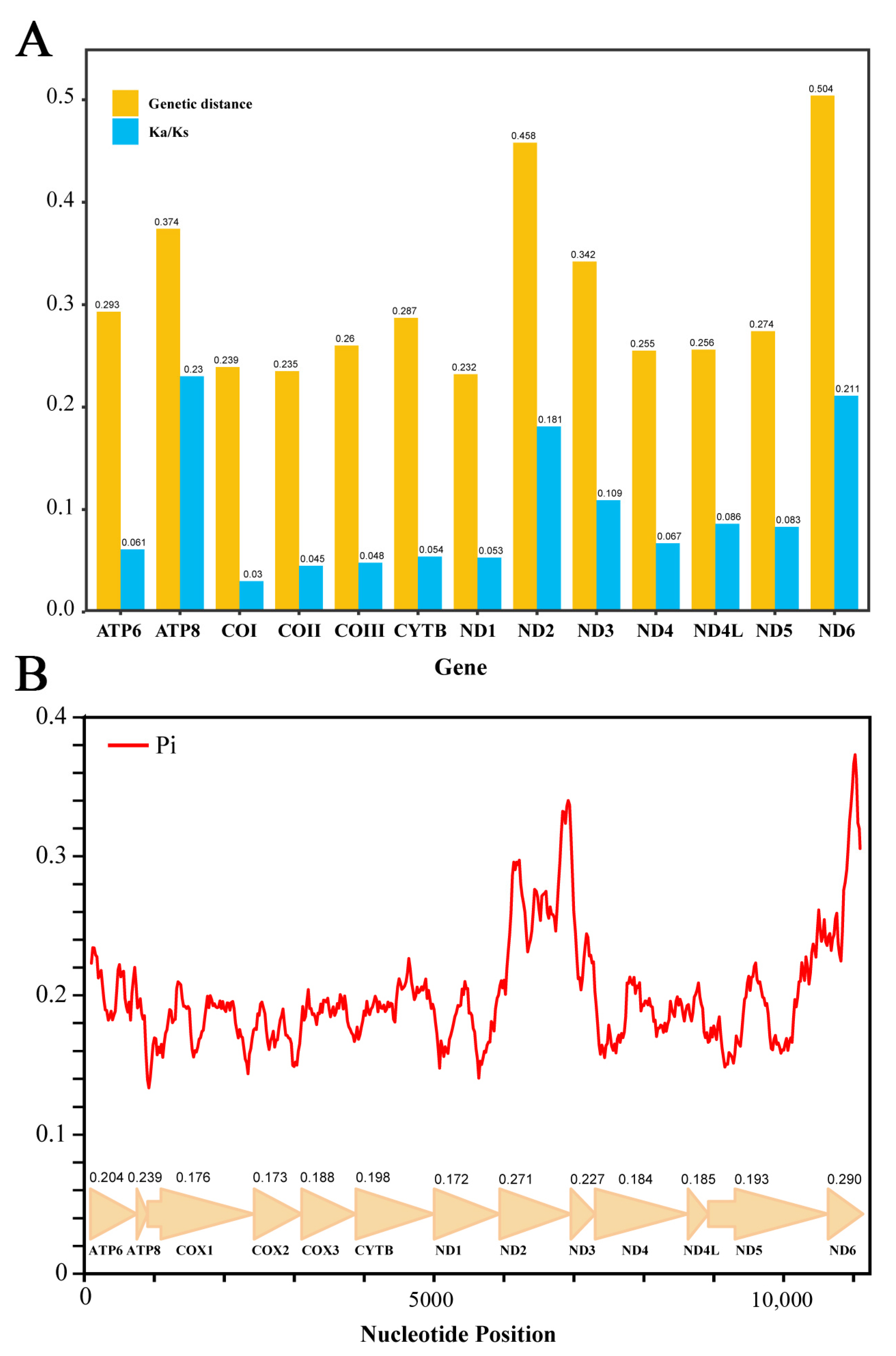
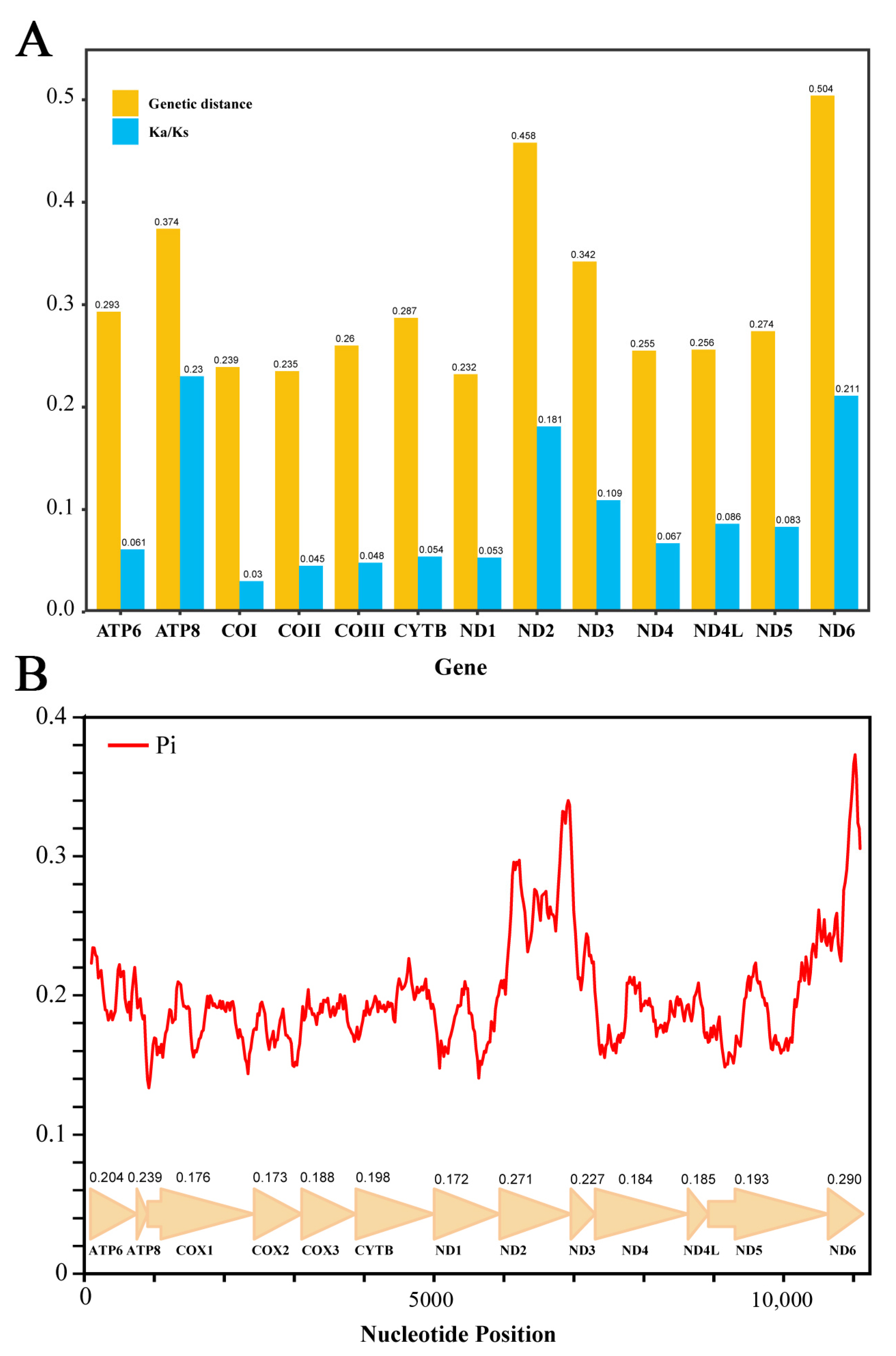
3.2. Analysis of Protein-Coding Genes
3.3. Transfer and Ribosomal RNA Genes
3.4. Non-Coding Regions
3.5. Gene Arrangement
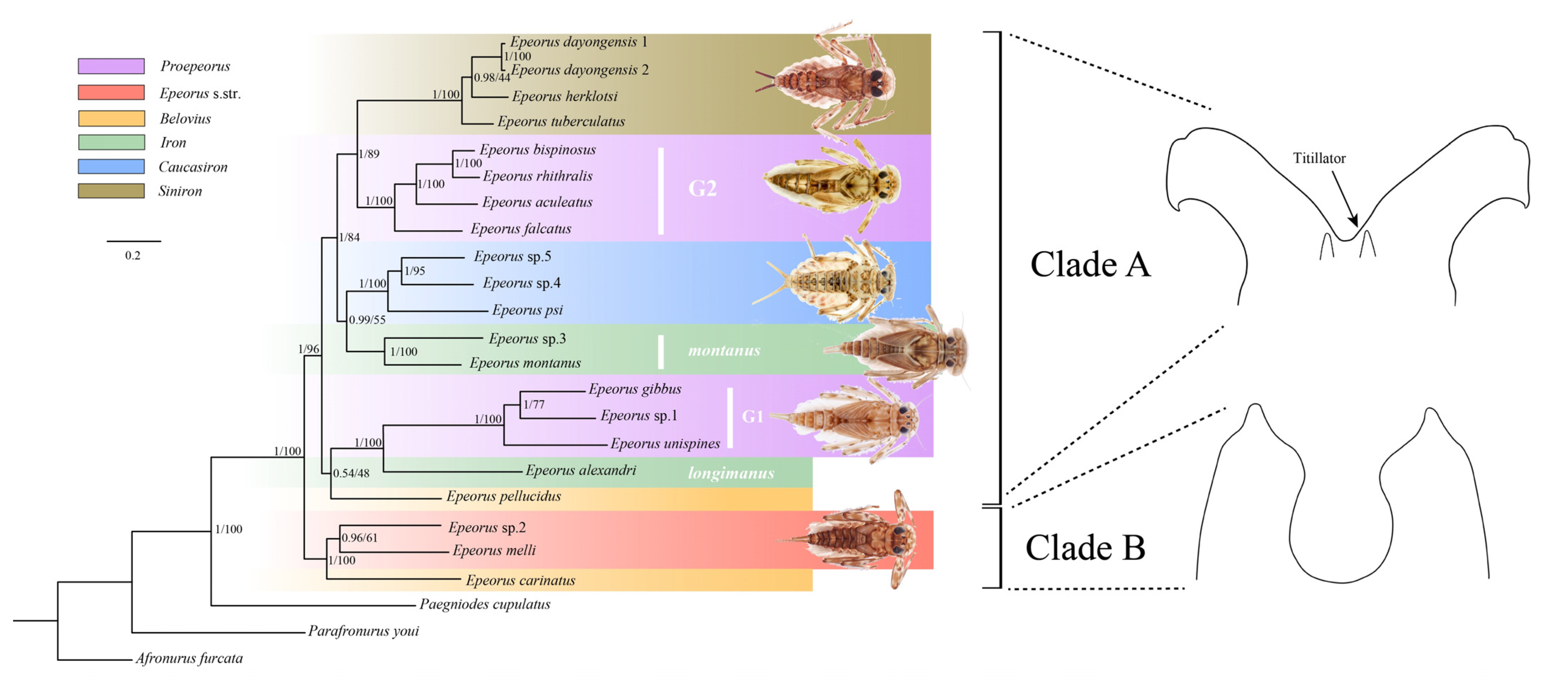
3.6. Phylogenetic Relationship
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ditsche-Kuru, P.; Koop, J.H.E. New insights into a life in current: Do the gill lamellae of Epeorus assimilis and Iron alpicola larvae (Ephemeroptera: Heptageniidae) function as a sucker or as friction pads? Aquat. Insects 2009, 31, 495–506. [Google Scholar] [CrossRef]
- Wang, T.Q.; McCafferty, W.P. Heptageniidae (Ephemeroptera) of the world. Part I: Phylogenetic higher classification. T. Am. Entomol. Soc. 2004, 130, 11–45. [Google Scholar]
- Webb, J.M.; McCafferty, W.P. Heptageniidae of the world. Part II. Key to the genera. Can. J. Arthropod Ident. 2008, 7, 1–55. [Google Scholar]
- Sartori, M.; Brittain, J.E. Order Ephemeroptera. In Ecology and General Biology: Thorp and Covich’s Freshwater Invertebrates; Thorp, J., Rogers, D.C., Eds.; Academic Press: Salt Lake City, UT, USA, 2015; pp. 873–891. [Google Scholar]
- Sivaruban, T.; Barathy, S.; Arunchalam, M.; Venkataraman, K.; Sivaramakrishnan, K.G. Epeorus petersi, a new species of Heptageniidae (Ephemeroptera) from the Western Ghats of southern India. Zootaxa 2013, 3731, 391–394. [Google Scholar] [CrossRef] [Green Version]
- Boonsoong, B.; Braasch, D. Heptageniidae (Insecta, Ephemeroptera) of Thailand. ZooKeys 2013, 272, 61–93. [Google Scholar] [CrossRef] [Green Version]
- Zhenxing, M.; Changfa, Z. A new subgenus of Epeorus and its five species from China (Ephemeroptera: Heptageniidae). Insect Syst. Evol. 2022, 53, 264–303. [Google Scholar] [CrossRef]
- Ma, Z.X.; Lei, Z.M.; Li, W.J.; Zhou, C.F. A revision of Epeorus (Proepeorus) in China, with descriptions of two new species (Ephemeroptera, Heptageniidae). J. Nat. Hist. 2021, 55, 1131–1159. [Google Scholar] [CrossRef]
- Braasch, D. Neue Eintagsfliegen der Gattungen Epeorus und Iron aus dem Himalaja (Ephemeroptera, Heptageniidae). Entomol. Nachr. Und Ber. 2006, 50, 79–88. [Google Scholar]
- Kluge, N.J. Phylogenetic System of Ephemeroptera; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; p. 442. [Google Scholar]
- Hrivniak, Ľ.; Sroka, P.; Bojková, J.; Godunko, R.J.; Soldán, T.; Staniczek, A.H. The impact of Miocene orogeny for the diversification of Caucasian Epeorus (Caucasiron) mayflies (Ephemeroptera: Heptageniidae). Mol. Phylogenet. Evol. 2020, 146, 106735. [Google Scholar] [CrossRef]
- Kluge, N.J.; Tiunova, T.M. Palearctic mayflies of the group longimanus of the subgenus Iron of Epeorus (Ephemeroptera, Heptageniidae). Vestn. Zool. 1989, 4, 7–14. [Google Scholar]
- Yan, L.; Xu, W.; Zhang, D.; Li, J. Comparative analysis of the mitochondrial genomes of flesh flies and their evolutionary implication. Int. J. Biol. Macromol. 2021, 174, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, G.; Itoh, H.; Fukuda, H.; Kojima, S. The complete mitochondrial genome of the sand bubbler crab Scopimera globosa and its phylogenetic position. Genomics 2021, 113, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhou, L.; Yang, W.T.; Miao, L.J.; Li, Z.; Zhang, X.J.; Wang, Y.; Gui, J.F. Comparative mitogenome analyses uncover mitogenome features and phylogenetic implications of the subfamily Cobitinae. BMC Genom. 2021, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Lei, Z.M.; Li, W.J.; Zhang, W.; Zhou, C.F. Comparative Mitogenomic Analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved Intergenic Spacer and tRNA Gene Duplication. Insects 2021, 12, 170. [Google Scholar] [CrossRef]
- Xu, X.D.; Guan, J.Y.; Zhang, Z.Y.; Cao, Y.R.; Cai, Y.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Insight into the phylogenetic relationships among three subfamilies within Heptageniidae (Insecta: Ephemeroptera) along with low-temperature selection pressure analyses using mitogenomes. Insects 2021, 12, 656. [Google Scholar] [CrossRef]
- Du, C.; Zhang, L.; Lu, T.; Ma, J.; Zeng, C.; Yue, B.; Zhang, X. Mitochondrial genomes of blister beetles (Coleoptera, Meloidae) and two large intergenic spacers in Hycleus genera. BMC Genom. 2017, 18, 698. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Ma, Z.X.; Zhou, C.F. Novel gene rearrangement pattern in the mitochondrial genomes of Torleya mikhaili and Cincticostella fusca (Ephemeroptera: Ephemerellidae). Int. J. Biol. Macromol. 2020, 165, 3106–3114. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Ma, Z.X.; Zhou, C.F. First complete mitogenomes of three mayflies in the genus Afronurus (Ephemeroptera: Heptageniidae) and their implications for phylogenetic reconstruction. Biologia 2021, 76, 2291–2302. [Google Scholar] [CrossRef]
- Xu, X.D.; Jia, Y.Y.; Cao, S.S.; Zhang, Z.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Six complete mitochondrial genomes of mayflies from three genera of Ephemerellidae (Insecta: Ephemeroptera) with inversion and translocation of trnI rearrangement and their phylogenetic relationships. PeerJ 2020, 8, e9740. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Wang, Y.Y.; Sun, J.Z.; Han, Y.K.; Zhou, C.F. The complete mitochondrial genome of Paegniodes cupulatus (Ephemeroptera: Heptageniidae). Mitochondrial DNA A 2016, 27, 925–926. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Zhang, S.S.; Zhang, L.P.; Yu, D.N.; Zhang, J.Y.; Cheng, H.Y. The complete mitochondrial genome of Epeorus herklotsi (Ephemeroptera: Heptageniidae) and its phylogeny. Mitochondrial DNA B 2018, 3, 303–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Li, R.; Zhou, C.F. Complete mitochondrial genomes of Epeorus carinatus and E. dayongensis (Ephemeroptera: Heptageniidae): Genomic comparison and phylogenetic inference. Gene 2021, 777, 145467. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Nicolas, D.; Patrick, M.; Guillaume, S. Novoplasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotech. 1994, 3, 294–299. [Google Scholar]
- Ogden, T.H.; Whiting, M.F. Phylogeny of Ephemeroptera (mayflies) based on molecular evidence. Mol. Phylogenet. Evol. 2005, 37, 625–643. [Google Scholar] [CrossRef]
- Yanai, Z.; Sartori, M.; Dor, R.; Dorchin, N. Molecular phylogeny and morphological analysis resolve a long-standing controversy over generic concepts in Ecdyonurinae mayflies (Ephemeroptera: Heptageniidae). Syst. Entomol. 2017, 42, 182–193. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Alejandro, S.G. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partition Finder 2: New methods for selecting partitioned models of evolution formolecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Wayne, P.; Terri, S. The CIPRES science gateway: A community resource for phylogenetic analyses. In Proceedings of the 2011 TeraGrid Conference: Extreme Digital Discovery, Salt Lake City, UT, USA, 18–21 July 2011; pp. 1–8. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Carmona, M.; Cameron, S.L.; Quiroga, C.F.P. How are the mitochondrial genomes reorganized in Hexapoda? Differential evolution and the first report of convergences within Hexapoda. Gene 2021, 791, 145719. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus | Subgenus | Species Group | Species | GenBank ID |
|---|---|---|---|---|---|
| Heptageniidae | Epeorus | Proepeorus | G1 | Epeorus gibbus | OK495692 (this study) |
| G1 | Epeorus unispinosus | OK495693 (this study) | |||
| G1 | Epeorus sp. 1 | OK495694 (this study) | |||
| G2 | Epeorus aculeatus | OK495695 (this study) | |||
| G2 | Epeorus bispinosus | OK495696 (this study) | |||
| G2 | Epeorus rhithralis | OK495697 (this study) | |||
| G2 | Epeorus falcatus | MW381293 | |||
| Epeorus | Epeorus melli | MW381294 | |||
| Epeorus sp. 2 | OK495698 (this study) | ||||
| Belovius | Epeorus carinatus | MT112896 | |||
| Epeorus pellucidus | MW381296 | ||||
| Iron | longimanus | Epeorus alexandri | OK495699 (this study) | ||
| montanus | Epeorus montanus | MW381295 | |||
| montanus | Epeorus sp. 3 | OK495700 (this study) | |||
| Siniron | Epeorus herklotsi | OK495701 (this study) | |||
| Epeorus dayongensis 01 | MT112895 | ||||
| Epeorus dayongensis 02 | OK495703 (this study) | ||||
| Epeorus tuberculatus | OK495702 (this study) | ||||
| Caucasiron | Epeorus psi | OK495704 (this study) | |||
| Epeorus sp. 4 | OK495705 (this study) | ||||
| Epeorus sp. 5 | OK495706 (this study) | ||||
| Out group | |||||
| Paegniodes | Paegniodes cupulatus | MW381300 | |||
| Parafronurus | Parafronurus youi | EU349015 | |||
| Afronurus | Afronurus furcata | MW381292 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Z.; Li, R.; Zhu, B.; Zheng, X.; Zhou, C. Comparative Mitogenome Analyses of Subgenera and Species Groups in Epeorus (Ephemeroptera: Heptageniidae). Insects 2022, 13, 599. https://doi.org/10.3390/insects13070599
Ma Z, Li R, Zhu B, Zheng X, Zhou C. Comparative Mitogenome Analyses of Subgenera and Species Groups in Epeorus (Ephemeroptera: Heptageniidae). Insects. 2022; 13(7):599. https://doi.org/10.3390/insects13070599
Chicago/Turabian StyleMa, Zhenxing, Ran Li, Binqing Zhu, Xuhongyi Zheng, and Changfa Zhou. 2022. "Comparative Mitogenome Analyses of Subgenera and Species Groups in Epeorus (Ephemeroptera: Heptageniidae)" Insects 13, no. 7: 599. https://doi.org/10.3390/insects13070599
APA StyleMa, Z., Li, R., Zhu, B., Zheng, X., & Zhou, C. (2022). Comparative Mitogenome Analyses of Subgenera and Species Groups in Epeorus (Ephemeroptera: Heptageniidae). Insects, 13(7), 599. https://doi.org/10.3390/insects13070599