
Global Profiling of Genes Expressed in the Silk Glands of Philippine-Reared Mulberry Silkworms (Bombyx mori)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
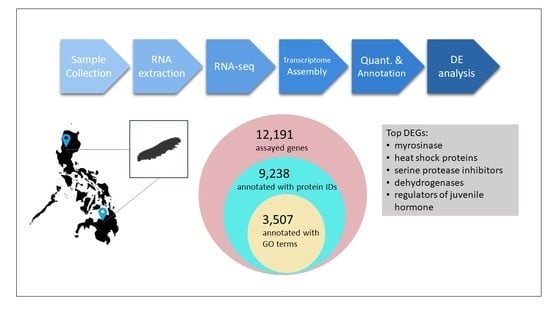
2.1. Insect and Tissue Collection
2.2. RNA Extraction and Sequencing
2.3. Preprocessing, Alignment, and Transcriptome Assembly
2.4. Expression Quantification and Differential Expression Analysis
2.5. Quantitative Real-Time PCR (qPCR)
3. Results
3.1. Selection of Representative Silkworm Strains
3.2. RNA Sequencing and Quality Control
3.3. RNA-seq Read Alignment to the Reference Genome
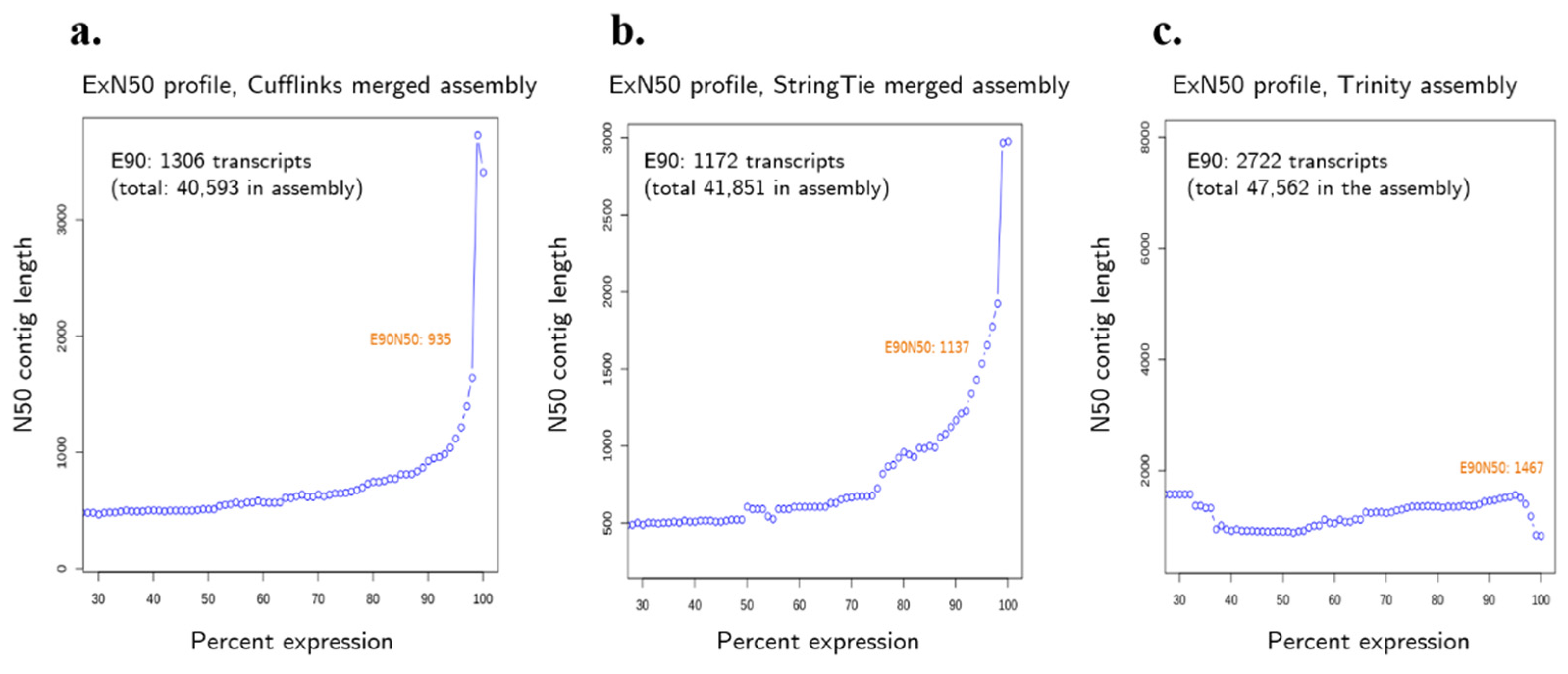
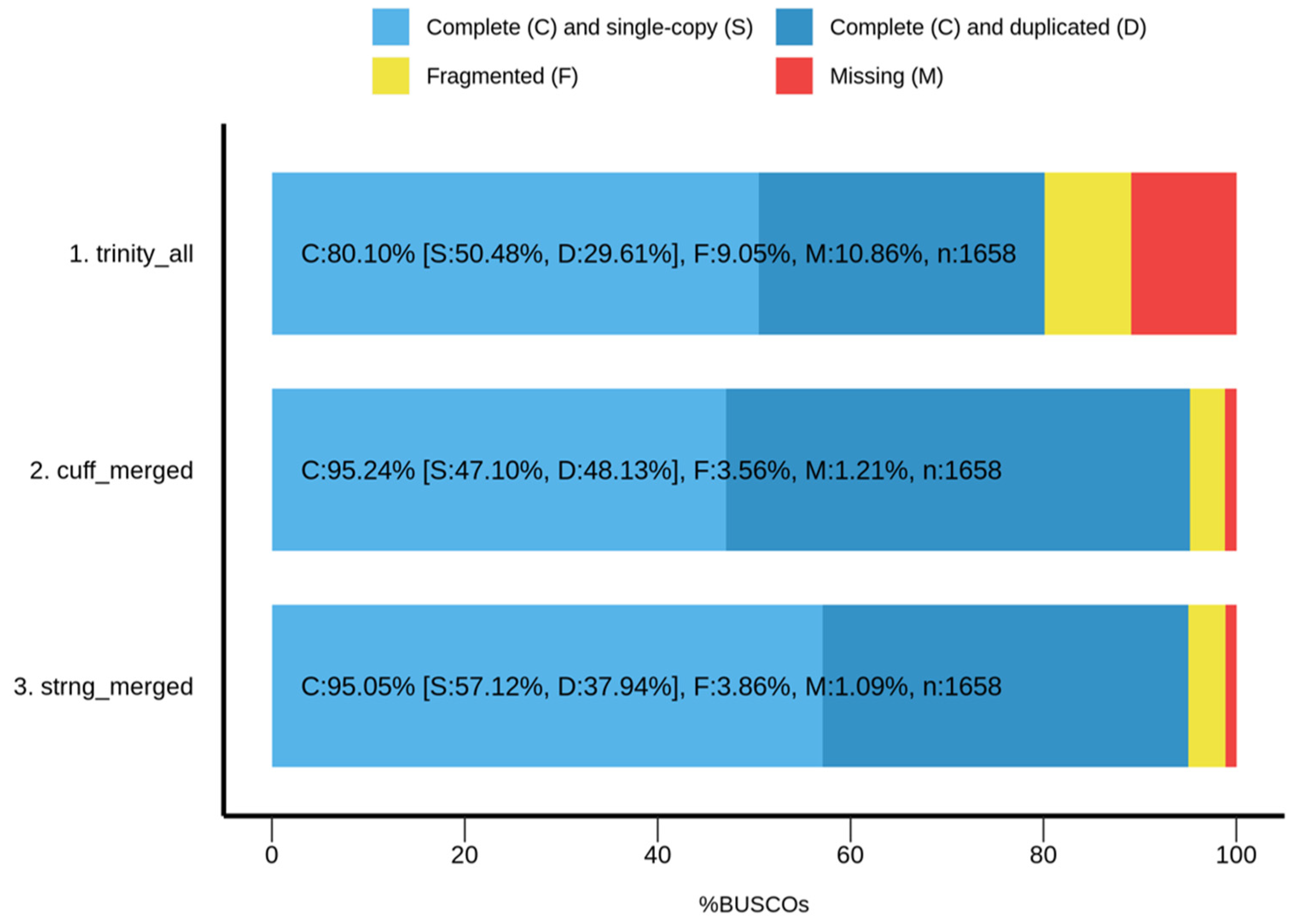
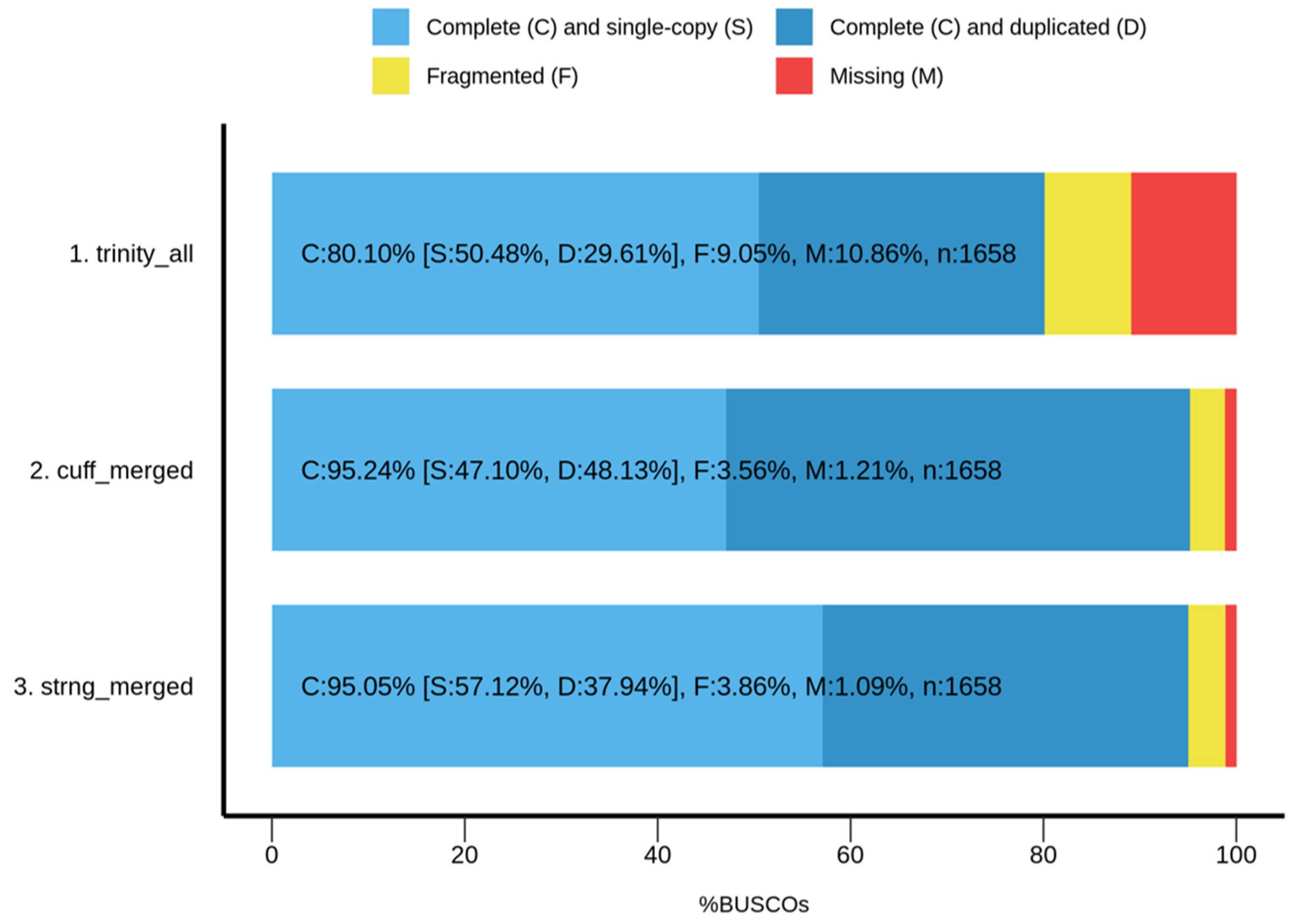
3.4. Transcriptome Assembly and Evaluation Metrics
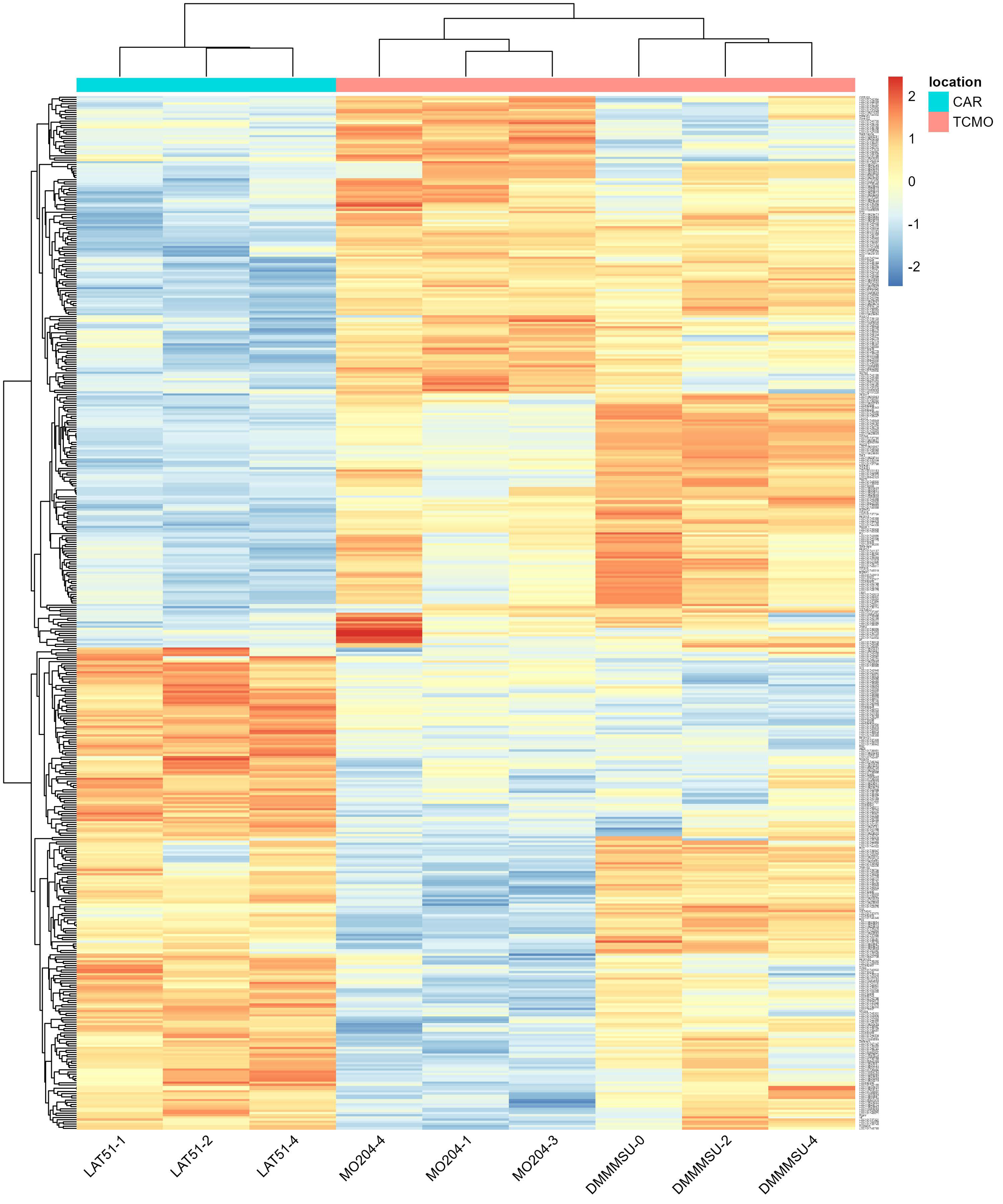
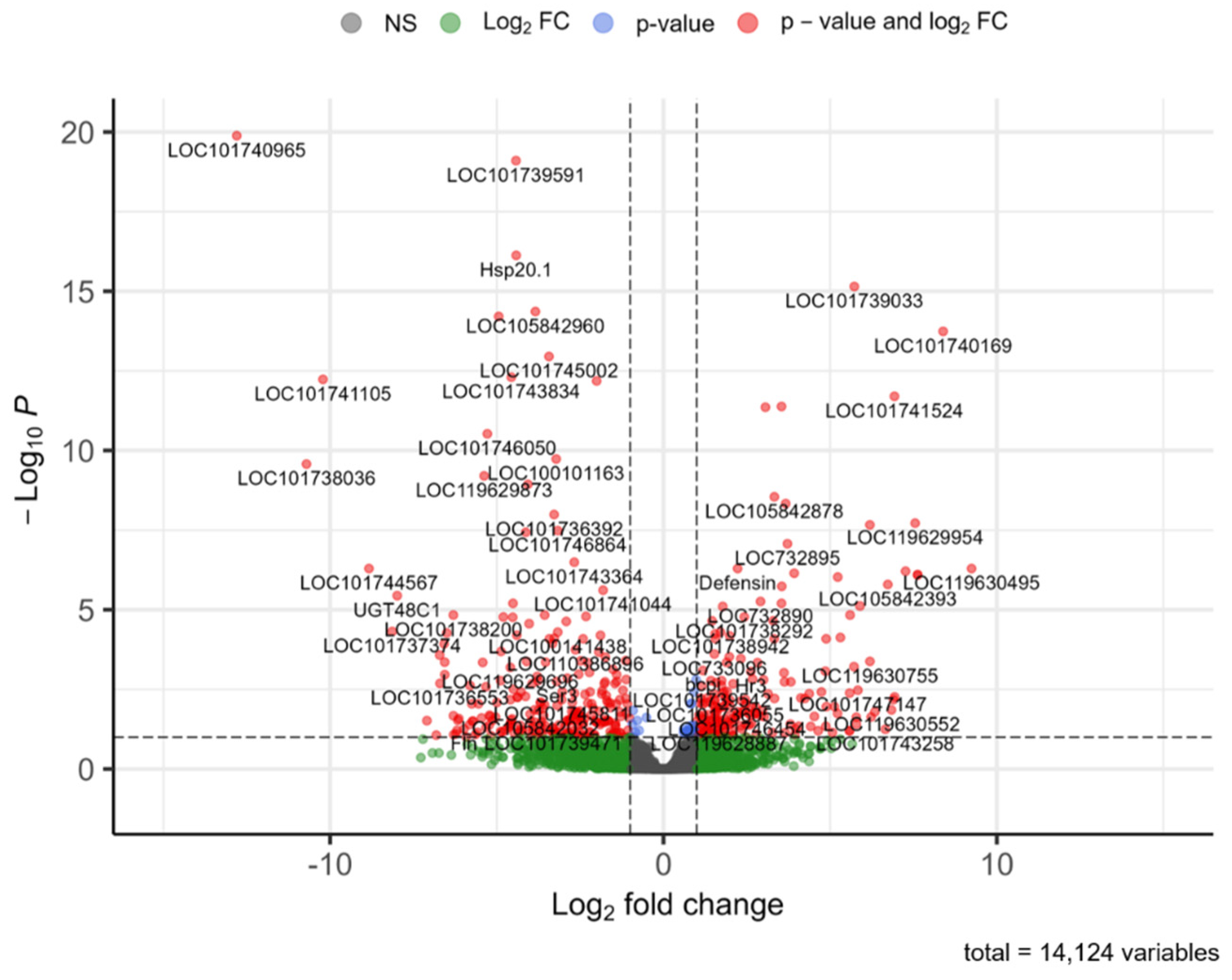
3.5. Expression Quantification, Normalization, and Differential Expression Analysis
3.6. Functional Annotation of Select DEGs
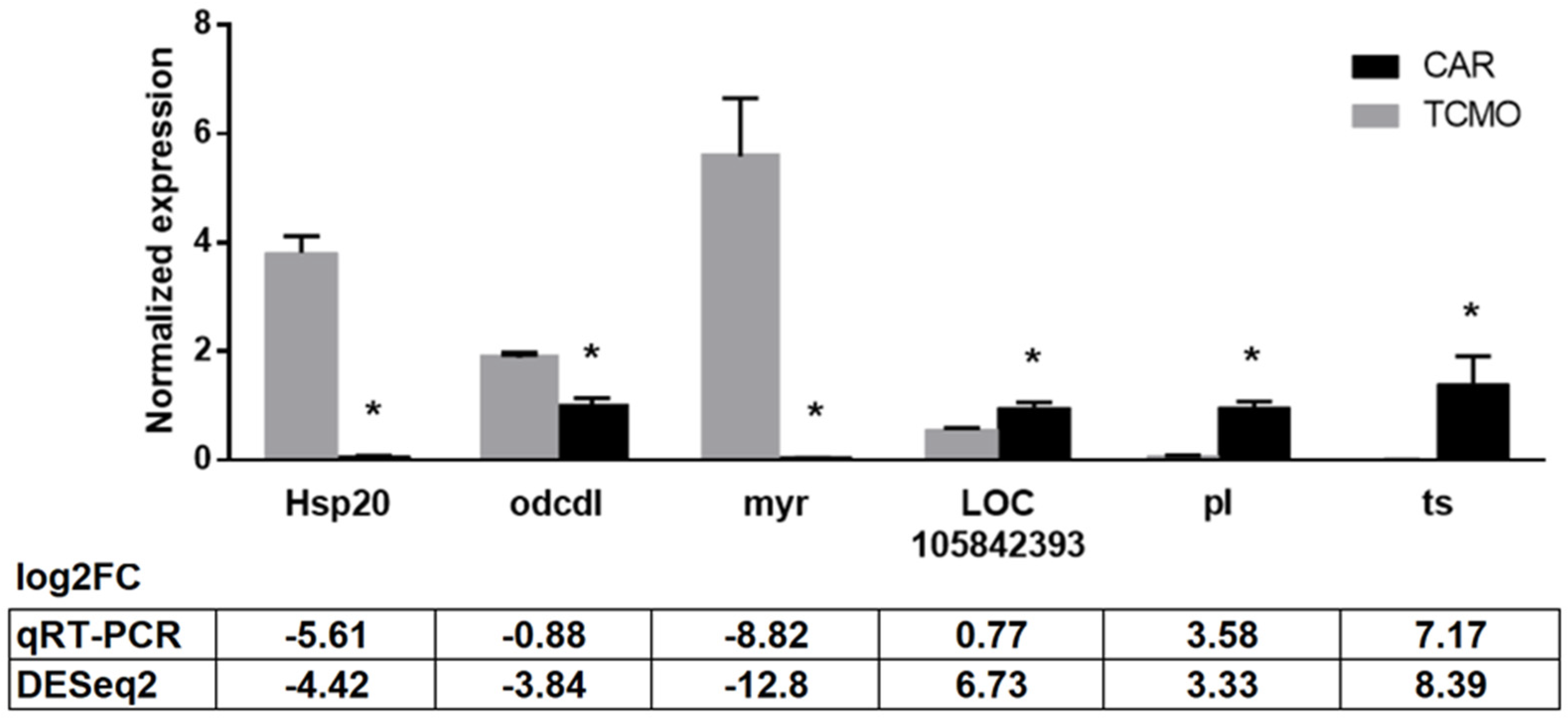
3.7. qPCR Validation of Select DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trevisan, A.; Hart, M.; Yuste, V.; Hjoellund, A.; Galle, D.; Bergman, O. Cocoon Silk: A Natural Architecture. Available online: https://web.archive.org/web/20120507085636/http://www.senature.com/research/publications/cocoon-silk-a-natural-architecture (accessed on 19 October 2021).
- Kunz, R.I.; Brancalhão, R.M.C.; de Ribeiro, L.F.C.; Natali, M.R.M. Silkworm Sericin: Properties and Biomedical Applications. BioMed Res. Int. 2016, 2016, 8175701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PhilFIDA. The Negros Silk Project. Available online: http://www.philfida.da.gov.ph/index.php/recent-news-articles/114-the-negros-silk-project (accessed on 4 November 2021).
- Zanatta, D.B.; Bravo, J.P.; Barbosa, J.F.; Munhoz, R.E.F.; Fernandez, M.A. Evaluation of Economically Important Traits from Sixteen Parental Strains of the Silkworm Bombyx Mori L (Lepidoptera: Bombycidae). Neotrop. Entomol. 2009, 38, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.C. Silkworm Genetics Research: Developing Silkworm Strains Adapted to Lowland Philippine Conditions. Presented at the Development Fund Professorial Chair Lecture, University of the Philippines Los Baños, College, Laguna. 2003. Available online: https://agris.fao.org/agris-search/search.do?recordID=PH2004000177 (accessed on 4 November 2021).
- Supsup, G.E.; Abuan, J.P.; Viduya, M.M.; Sanchez, M.L.; Ulat, M.E. Selection of Superior Bivoltine Silkworm Hybrids through Combining Ability Test and Multiple Traits Evaluation Index. IAMURE Int. J. Ecol. Conserv. 2018, 23, 1. [Google Scholar]
- Velasco, A.B.; Bacuso, P.M.; Cabrito, F.P.; Fernandez, R.W.; Alvarez, V.B.; de Guzman, Z.I.; Villanueva, E.P. New Breeds of Filipino Silkworm Varieties Commercialized. Philipp. Technol. J. 1994, 19. Available online: https://agris.fao.org/agris-search/search.do?recordID=PH9710645 (accessed on 4 November 2021).
- Aro, S.C. Silk Production on the Decline. Sunstar. 2015. Available online: https://web.archive.org/web/20220722062934/https://www.sunstar.com.ph//article/40526 (accessed on 22 July 2022).
- Villanueva, R.C. Problems and Issues Affecting the Pace of Sericulture Development in the Philippines. Don Marian. Marcos Meml. State Univ. Res. Ext. J. Bacnotan Campus. 1999. Available online: https://agris.fao.org/agris-search/search.do?recordID=PH2007M00034 (accessed on 20 June 2022).
- Li, J.; Qin, S.; Yu, H.; Zhang, J.; Liu, N.; Yu, Y.; Hou, C.; Li, M. Comparative Transcriptome Analysis Reveals Different Silk Yields of Two Silkworm Strains. PLoS ONE 2016, 11, e0155329. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.-M.; Hu, B.-L.; Zhou, Q.-Z.; Yu, Q.-Y.; Zhang, Z. Comparative Analysis of the Silk Gland Transcriptomes between the Domestic and Wild Silkworms. BMC Genom. 2015, 16, 60. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Wei, Z.; Luo, Y.; Guo, H.; Zhang, G.; Xia, Q.; Wang, Y. SilkDB 3.0: Visualizing and Exploring Multiple Levels of Data for Silkworm. Nucleic Acids Res. 2020, 48, D749–D755. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, M.; Minami, H.; Suetsugu, Y.; Ohyanagi, H.; Satoh, C.; Antonio, B.; Nagamura, Y.; Kadono-Okuda, K.; Kajiwara, H.; Sezutsu, H.; et al. KAIKObase: An Integrated Silkworm Genome Database and Data Mining Tool. BMC Genom. 2009, 10, 486. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, G.; Tian, J.; Liu, H.; Yang, H.; Yi, Y.; Wang, J.; Shi, X.; Jiang, F.; Yao, B.; et al. Transcriptome Analysis of the Silkworm (Bombyx Mori) by High-Throughput RNA Sequencing. PLoS ONE 2012, 7, e43713. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.D.; Muthulakshmi, M.; Arunkumar, K.P.; Madhu, M.; Sreenu, V.B.; Pavithra, V.; Bose, B.; Nagarajaram, H.A.; Mita, K.; Shimada, T.; et al. SilkSatDb: A Microsatellite Database of the Silkworm, Bombyx Mori. Nucleic Acids Res. 2005, 33, D403–D406. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.-E.; Zhang, H.-H.; Xia, T.; Han, M.-J.; Shen, Y.-H.; Zhang, Z. BmTEdb: A Collective Database of Transposable Elements in the Silkworm Genome. Database 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Li, S.; Feng, Q. Advances in Silkworm Studies Accelerated by the Genome Sequencing of Bombyx Mori. Annu. Rev. Entomol. 2014, 59, 513–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Zhang, R.; Cao, Y.-Y.; Du, J.; Thakur, K.; Tang, S.-M.; Hu, F.; Wei, Z.-J. Transcriptome Analysis Reveals the Gene Expression Changes in the Silkworm (Bombyx Mori) in Response to Hydrogen Sulfide Exposure. Insects 2021, 12, 1110. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Tsubota, T.; Jouraku, A.; Sezutsu, H.; Bono, H. Reference Transcriptome Data in Silkworm Bombyx Mori. Insects 2021, 12, 519. [Google Scholar] [CrossRef]
- The International Silkworm Genome Consortium. The Genome of a Lepidopteran Model Insect, the Silkworm Bombyx Mori. Insect Biochem. Mol. Biol. 2008, 38, 1036–1045. [Google Scholar] [CrossRef]
- Dundar, W.F.; Skrabanek, L.; Zumbo, P. Introduction to Differential Gene Expression Analysis Using RNA-Seq 2019. Available online: https://chagall.med.cornell.edu/RNASEQcourse/Intro2RNAseq.pdf (accessed on 2 December 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Haas, B. Transcriptome Contig Nx and ExN50 Stats Trinityrnaseq/Trinityrnaseq Wiki. Available online: https://github.com/trinityrnaseq/trinityrnaseq (accessed on 2 December 2021).
- Li, B.; Fillmore, N.; Bai, Y.; Collins, M.; Thomson, J.A.; Stewart, R.; Dewey, C.N. Evaluation of de Novo Transcriptome Assemblies from RNA-Seq Data. Genome Biol. 2014, 15, 553. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 2 December 2021).
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Rahmathulla, V.K. Management of Climatic Factors for Successful Silkworm (Bombyx Mori L.) Crop and Higher Silk Production: A Review. Psyche 2012, 2012, e121234. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.S.; Basavaraja, H.K.; Kumar, C.M.K.; Reddy, N.M.; Datta, R.K. On the Breeding of “CSR18xCSR19”- A Robust Bivoltine Hybrid of Silkworm, Bombyx Mori L. for the Tropics. Int. J. Ind. Entomol. 2002, 5, 153–162. [Google Scholar]
- Guo, H.; Huang, C.; Jiang, L.; Cheng, T.; Feng, T.; Xia, Q. Transcriptome Analysis of the Response of Silkworm to Drastic Changes in Ambient Temperature. Appl. Microbiol. Biotechnol. 2018, 102, 10161–10170. [Google Scholar] [CrossRef]
- Zahn-Zabal, M.; Michel, P.-A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; Rech de Laval, V.; et al. The NeXtProt Knowledgebase in 2020: Data, Tools and Usability Improvements. Nucleic Acids Res. 2020, 48, D328–D334. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.-F.; Li, Y.-N.; Yi, Y.-Z.; Xiao, X.-G.; Zhang, Z.-F. Identification and Characterization of 30 K Protein Genes Found in Bombyx Mori (Lepidoptera: Bombycidae) Transcriptome. J. Insect Sci. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.K.; Pakkianathan, B.C.; Kumar, M.; Prasad, T.; Kannan, M.; König, S.; Krishnan, M. Vitellogenin from the Silkworm, Bombyx Mori: An Effective Anti-Bacterial Agent. PLoS ONE 2013, 8, e73005. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, M.; Su, J.; Ma, H.; Zheng, X.; Li, Q.; Shi, S.; Qin, L. Identification and Characterization of a Novel Microvitellogenin from the Chinese Oak Silkworm Antheraea Pernyi. PLoS ONE 2015, 10, e0131751. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Fässler, R.; Hohenester, E. Laminin: The Crux of Basement Membrane Assembly. J. Cell Biol. 2004, 164, 959–963. [Google Scholar] [CrossRef] [Green Version]
- Daillie, J. Juvenile Hormone Modifies Larvae and Silk Gland Development in Bombyx Mori. Biochimie 1979, 61, 275–281. [Google Scholar] [CrossRef]
- Zhao, X.-M.; Liu, C.; Jiang, L.-J.; Li, Q.-Y.; Zhou, M.-T.; Cheng, T.-C.; Mita, K.; Xia, Q.-Y. A Juvenile Hormone Transcription Factor Bmdimm-Fibroin H Chain Pathway Is Involved in the Synthesis of Silk Protein in Silkworm, Bombyx Mori *. J. Biol. Chem. 2015, 290, 972–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Coll, M.; Applebaum, S.W. Effect of Temperature and Photoperiod on Juvenile Hormone Biosynthesis and Sexual Maturation in the Cotton Bollworm, Helicoverpa Armigera: Implications for Life History Traits. Insect Biochem. Mol. Biol. 2000, 30, 863–868. [Google Scholar] [CrossRef]
- Liu, Y.; Henderson, G.; Mao, L.; Laine, R.A. Effects of Temperature and Nutrition on Juvenile Hormone Titers of Coptotermes Formosanus (Isoptera: Rhinotermitidae). Ann. Entomol. Soc. Am. 2005, 98, 732–737. [Google Scholar] [CrossRef]
- Geister, T.L.; Lorenz, M.W.; Meyering-Vos, M.; Klaus, H.H.; Fischer, K. Effects of Temperature on Reproductive Output, Egg Provisioning, Juvenile Hormone and Vitellogenin Titres in the Butterfly Bicyclus Anynana. J. Insect Physiol. 2008, 54, 1253–1260. [Google Scholar] [CrossRef]
- Gupta, A.B.; Wee, L.E.; Zhou, Y.T.; Hortsch, M.; Low, B.C. Cross-Species Analyses Identify the BNIP-2 and Cdc42GAP Homology (BCH) Domain as a Distinct Functional Subclass of the CRAL_TRIO/Sec14 Superfamily. PLoS ONE 2012, 7, e33863. [Google Scholar] [CrossRef]
- Ailion, M.; Thomas, J.H. Isolation and Characterization of High-Temperature-Induced Dauer Formation Mutants in Caenorhabditis Elegans. Genetics 2003, 165, 127–144. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences (5′ to 3′) Forward Reverse | Product Length (bp) | Protein ID |
|---|---|---|---|
| Downregulated genes (CAR vs. TCMO) | |||
| Hsp 20 | GCCAACGATGTCCAGAGATT CTGCCTCTCCTCGTGCTTAC | 196 | heat shock protein hsp20.1 |
| odcdl | AAATGTCTGGGTGGAAGCAG GGGTGGAATCATCAGGTTGT | 235 | oxygen-dependent choline dehydrogenase-like |
| myr | GGAGACCGAGTGAAGACCTG GCTGTGGCATGAGCTAACAA | 152 | myrosinase 1 |
| Upregulated genes (CAR vs. TCMO) | |||
| pl | TGTTGTCGCTTTTCATCTGC AGTTTTTGAGCGCCGTATTG | 207 | peroxidase-like |
| LOC105842393 | AAATGGCGCACAAATAGAGG CCAAGCTCTGCGTAAGGTTC | 177 | uncharacterized protein |
| ts | AAACCGAGCGTCCACTTATG TATTGTGATGGCAGCGGTAA | 186 | uncharacterized protein (tetraspanin family) |
| Housekeeping gene for normalization | |||
| rp49 | CAGGCGGTTCAAGGGTCAATAC TGCTGGGCTCTTTCCACGA | 213 | ribosomal protein 49 |
| Strain | Origin | Strain Classification | Cocoon | Cocoon Shell Percentage | Raw Silk Percentage |
|---|---|---|---|---|---|
| MO204 | TCMO | Priority | Oval, white | 22.92% | 22.42% |
| DMMMSU119 | TCMO | Poor performing | Peanut, white | 21.81% | 21.12% |
| LAT51 | CAR | Priority | Peanut, white | 21.57% | 17.44% |
| ITA | CAR | Priority | Oval, yellow gold | 20.27% | no data |
| Sample | Read Count | % ≥ Q30 | Mean Q Score | GC Content (%) |
|---|---|---|---|---|
| MO204-1 | 17,531,032 | 94.11 | 34.60 | 41 |
| MO204-3 | 8,590,562 | 93.49 | 34.47 | 43 |
| MO204-4 | 13,085,562 | 94.10 | 34.59 | 44 |
| DMMMSU-0 | 11,406,238 | 92.68 | 34.29 | 46 |
| DMMMSU-2 | 21,789,720 | 92.38 | 34.22 | 46 |
| DMMMSU-4 | 15,464,914 | 91.51 | 34.04 | 46 |
| LAT51-1 | 12,321,992 | 93.70 | 34.51 | 42 |
| LAT51-2 | 22,767,422 | 93.91 | 34.56 | 41 |
| LAT51-4 | 8,793,834 | 94.74 | 34.73 | 44 |
| ITA-1 | 3,621,664 | 86.80 | 33.05 | 48 |
| ITA-2 | 4,633,324 | 88.96 | 33.51 | 48 |
| ITA-3 | 3106 | 65.36 | 28.55 | 75 |
| Trinity | Cufflinks | StringTie | |
|---|---|---|---|
| Total Trinity “genes” | 47,562 | 40,593 | 41,851 |
| Total Trinity transcripts | 69,948 | 40,593 | 41,851 |
| Percent GC | 38.12% | 42.08% | 41.66% |
| N10 | 3257 | 7944 | 7333 |
| N20 | 2487 | 6108 | 5595 |
| N30 | 1992 | 4987 | 4491 |
| N40 | 1635 | 4126 | 3640 |
| N50 | 1313 | 3414 | 2981 |
| Median contig length | 468 | 1738 | 1226 |
| Average contig length | 800.21 | 2305.35 | 1789.3 |
| Assembly | RSEM-Eval | Nucleotide F1 (Unweighted) | Contig F1 (Unweighted) | Weighted k-Mer Recall | k-Mer Compression Score |
|---|---|---|---|---|---|
| Trinity | −8.35 × 109 | 0.38019 | 0.00239295 | 0.678414 | −7.32371 |
| Cufflinks | −1.34 × 1010 | 0.682955 | 0.690038 | 0.545637 | −22.5079 |
| StringTie | −1.55 × 1010 | 0.775391 | 0.677147 | 0.545727 | −17.3472 |
| Downregulated Genes (CAR vs. TCMO) | ||||
|---|---|---|---|---|
| Gene ID | log2FC | padj | Protein ID | Function |
| LOC101740965 | −12.8030 | 1.31 × 10−20 | myrosinase 1 isoform X2 | hydrolysis of glucosinolates, defense enzyme |
| Hsp20.1 | −4.4182 | 7.45 × 10−17 | heat shock protein hsp20.1 | chaperone protein |
| LOC101739591 | −4.4249 | 7.92 × 10−20 | transient receptor potential channel pyrexia isoform X1 | ion transport, contains ankyrin repeat |
| LOC101741105 | −10.2153 | 5.85 × 10−13 | myrosinase 1 | hydrolysis of glucosinolates, defense enzyme |
| LOC101738036 | −10.7102 | 2.66 × 10−10 | serine protease inhibitor swm-1 | serine protease inhibitor, trypsin inhibitor |
| LOC105842960 | −3.8422 | 4.35 × 10−15 | oxygen-dependent choline dehydrogenase-like | oxidoreductase activity, FAD/NAD(P)-binding |
| Upregulated genes (CAR vs. TCMO) | ||||
| Gene ID | log2FC | padj | Protein ID | Function |
| LOC101739033 | 5.7239 | 7.09 × 10−16 | zinc finger protein 664 | dimer formation, metal binding |
| LOC101740169 | 8.3899 | 1.81 × 10−14 | uncharacterized protein LOC101740169 | tetraspanin family, scaffolding, cell adhesion, proliferation |
| LOC101741524 | 6.9289 | 1.99 × 10−12 | synaptic vesicle glycoprotein 2C | sugar transporter |
| LOC105842878 | 3.3250 | 2.85 × 10−9 | peroxidase-like | heme peroxidase, redox activity, oxidoreductase activity, defense enzyme |
| LOC119629954 | 7.5484 | 1.91 × 10−8 | zinc finger BED domain-containing protein 4-like isoform X2 | contains hAT dimerization domain, ribonuclease H-like superfamily: nucleotide metabolism and transport |
| LOC119630495 | 9.2422 | 5.08 × 10−7 | uncharacterized protein LOC119630495 | contains motifs: peptidase, Hsp70, TetR |
| LOC105842393 | 6.7307 | 1.61 × 10−6 | uncharacterized protein LOC105842393 | contains motifs: DUF5641 (pfam18701), unknown function, found in retrotransposons |
| Defensin | 2.2251 | 5.08 × 10−7 | defensin like protein 2 precursor | contains signal peptide |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Peña, P.N.O.; Lao, A.G.D.; Bautista, M.A.M. Global Profiling of Genes Expressed in the Silk Glands of Philippine-Reared Mulberry Silkworms (Bombyx mori). Insects 2022, 13, 669. https://doi.org/10.3390/insects13080669
de la Peña PNO, Lao AGD, Bautista MAM. Global Profiling of Genes Expressed in the Silk Glands of Philippine-Reared Mulberry Silkworms (Bombyx mori). Insects. 2022; 13(8):669. https://doi.org/10.3390/insects13080669
Chicago/Turabian Stylede la Peña, Pauline Nicole O., Adria Gabrielle D. Lao, and Ma. Anita M. Bautista. 2022. "Global Profiling of Genes Expressed in the Silk Glands of Philippine-Reared Mulberry Silkworms (Bombyx mori)" Insects 13, no. 8: 669. https://doi.org/10.3390/insects13080669
APA Stylede la Peña, P. N. O., Lao, A. G. D., & Bautista, M. A. M. (2022). Global Profiling of Genes Expressed in the Silk Glands of Philippine-Reared Mulberry Silkworms (Bombyx mori). Insects, 13(8), 669. https://doi.org/10.3390/insects13080669