
The Role of Feeding Characteristics in Shaping Gut Microbiota Composition and Function of Ensifera (Orthoptera)

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. DNA Library Construction
2.3. Metagenomics Data Assembly and Analysis
2.4. Statistical Analyses
3. Results
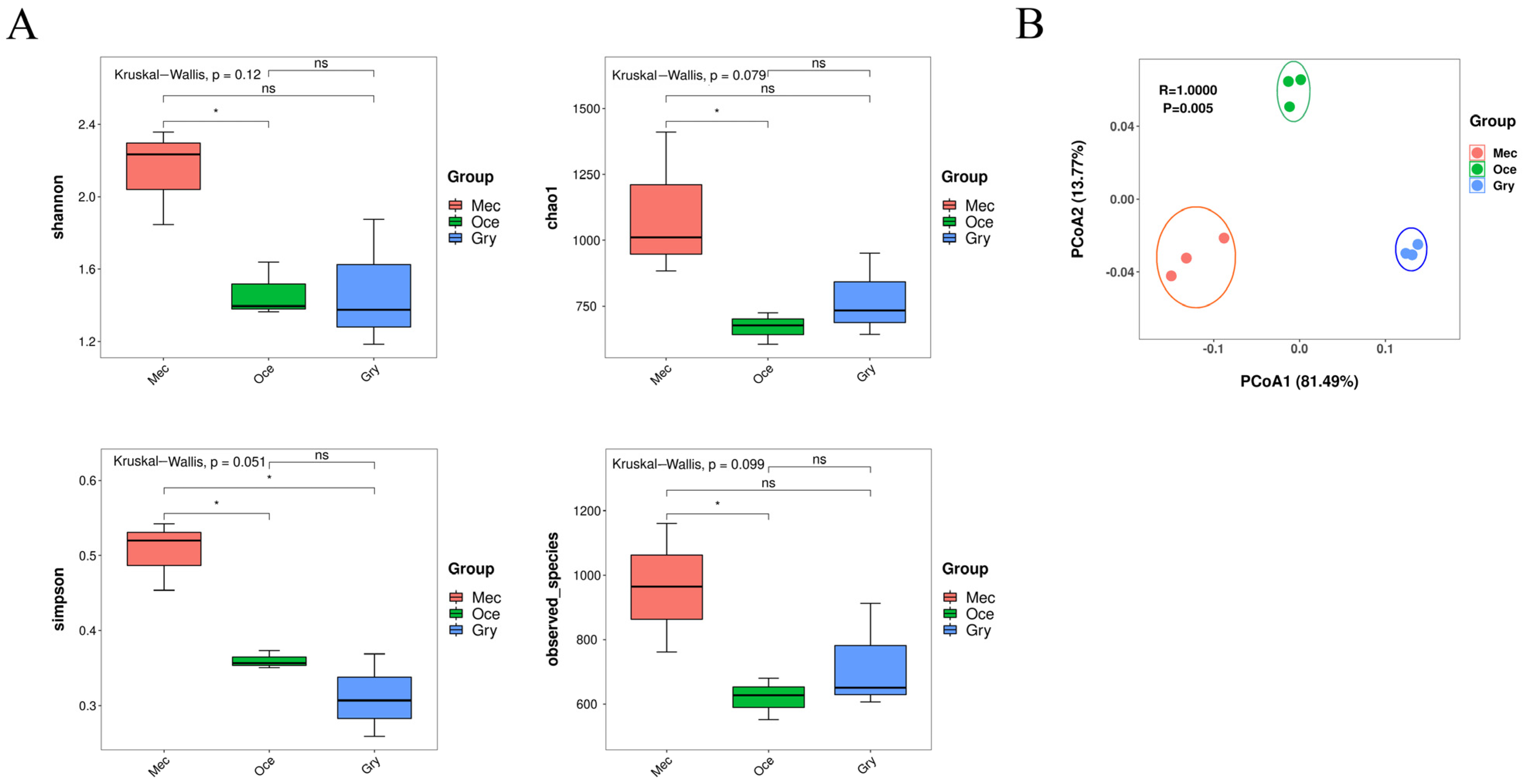
3.1. The Diversity and Composition of the Gut Microbiota
3.2. The Characteristics of the Gut Microbiota
3.3. Metabolic Potential Functions of Gut Microbiota According to the KEGG Database
3.4. Metabolic Potential Functions of Gut Microbiota According to CAZymes Database
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basset, Y.; Cizek, L.; Cuenoud, P.; Didham, R.K.; Guilhaumon, F.; Missa, O.; Novotny, V.; Odegaard, F.; Roslin, T.; Schmidl, J.; et al. Arthropod diversity in a tropical forest. Science 2012, 338, 1481–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Kim, M.S.; Park, D.S.; Yoon, C.; Nam, Y.D.; Kim, Y.J.; Choi, J.H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Ding, S.-Y.; Yuan, J.S. Comparison of insect gut cellulase and xylanase activity across different insect species with distinct food sources. BioEnergy Res. 2010, 4, 1–10. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yu, T.; Xie, S.; Du, K.; Liang, X.; Lan, Y.; Sun, C.; Lu, X.; Shao, Y. Comparative shotgun metagenomic data of the silkworm bombyx mori gut microbiome. Sci. Data 2018, 5, 180285. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. How do gut microbes help herbivores? Counting the ways. Science 2017, 355, 236. [Google Scholar] [CrossRef]
- Vogel, H.; Shukla, S.P.; Engl, T.; Weiss, B.; Fischer, R.; Steiger, S.; Heckel, D.G.; Kaltenpoth, M.; Vilcinskas, A. The digestive and defensive basis of carcass utilization by the burying beetle and its microbiota. Nat. Commun. 2017, 8, 15186. [Google Scholar] [CrossRef] [Green Version]
- Dillon, R.J.; Dillon, V.M. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 2004, 49, 71–92. [Google Scholar] [CrossRef]
- Nadarasah, G.; Stavrinides, J. Insects as alternative hosts for phytopathogenic bacteria. FEMS Microbiol. Rev. 2011, 35, 555–575. [Google Scholar] [CrossRef]
- Lavy, O.; Gophna, U.; Gefen, E.; Ayali, A. The effect of density-dependent phase on the locust gut bacterial composition. Front. Microbiol. 2018, 9, 3020. [Google Scholar] [CrossRef] [Green Version]
- Brumfield, K.D.; Huq, A.; Colwell, R.R.; Olds, J.L.; Leddy, M.B. Microbial resolution of whole genome shotgun and 16s amplicon metagenomic sequencing using publicly available neon data. PLoS ONE 2020, 15, e0228899. [Google Scholar]
- Huang, S.; Sheng, P.; Zhang, H. Isolation and identification of cellulolytic bacteria from the gut of holotrichia parallela larvae (coleoptera: Scarabaeidae). Int. J. Mol. Sci. 2012, 13, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
- Kwong, W.K.; Medina, L.A.; Koch, H.; Sing, K.W.; Soh, E.J.Y.; Ascher, J.S.; Jaffe, R.; Moran, N.A. Dynamic microbiome evolution in social bees. Sci. Adv. 2017, 3, e1600513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen-Vercoe, E. Bringing the gut microbiota into focus through microbial culture: Recent progress and future perspective. Curr. Opin. Microbiol. 2013, 16, 625–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Handelsman, J. Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 2003, 14, 303–310. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef] [Green Version]
- Yen, S.; Johnson, J.S. Metagenomics: A path to understanding the gut microbiome. Mamm. Genome 2021, 32, 282–296. [Google Scholar] [CrossRef]
- Han, D.; Gao, P.; Li, R.; Tan, P.; Xie, J.; Zhang, R.; Li, J. Multicenter assessment of microbial community profiling using 16s rrna gene sequencing and shotgun metagenomic sequencing. J. Adv. Res. 2020, 26, 111–121. [Google Scholar] [CrossRef]
- Beaudry, M.S.; Wang, J.; Kieran, T.J.; Thomas, J.; Bayona-Vasquez, N.J.; Gao, B.; Devault, A.; Brunelle, B.; Lu, K.; Wang, J.S.; et al. Improved microbial community characterization of 16s rrna via metagenome hybridization capture enrichment. Front. Microbiol. 2021, 12, 644662. [Google Scholar] [CrossRef]
- Narayan, N.R.; Weinmaier, T.; Laserna-Mendieta, E.J.; Claesson, M.J.; Shanahan, F.; Dabbagh, K.; Iwai, S.; DeSantis, T.Z. Piphillin predicts metagenomic composition and dynamics from dada2-corrected 16s rdna sequences. BMC Genom. 2020, 21, 56. [Google Scholar]
- Enagbonma, B.J.; Aremu, B.R.; Babalola, O.O. Profiling the functional diversity of termite mound soil bacteria as revealed by shotgun sequencing. Genes 2019, 10, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, I.J.; Rees, E.R.; Ross, J.; Miller, I.; Baxa, J.; Lopera, J.; Kerby, R.L.; Rey, F.E.; Kwan, J.C. Autometa: Automated extraction of microbial genomes from individual shotgun metagenomes. Nucleic Acids Res. 2019, 47, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, D.; Jhang, S.Y.; Kim, H.; Kim, S.Y.; Kwak, H.S.; Kim, S.H.; Lee, W.; Park, S.; Kim, H.; Cho, S.; et al. Accurate and strict identification of probiotic species based on coverage of whole-metagenome shotgun sequencing data. Front. Microbiol. 2019, 10, 1683. [Google Scholar] [CrossRef] [Green Version]
- Santo Domingo, J.W.; Kaufman, M.G.; Klug, M.J.; Tiedje, J.M. Characterization of the cricket hindgut microbiota with fluorescently labeled rrna-targeted oligonucleotide probes. Appl. Environ. Microbiol. 1998, 64, 752–755. [Google Scholar] [CrossRef] [Green Version]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Otani, S.; Mikaelyan, A.; Nobre, T.; Hansen, L.H.; Kone, N.A.; Sorensen, S.J.; Aanen, D.K.; Boomsma, J.J.; Brune, A.; Poulsen, M. Identifying the core microbial community in the gut of fungus-growing termites. Mol. Ecol. 2014, 23, 4631–4644. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cobas, A.E.; Maiques, E.; Angelova, A.; Carrasco, P.; Moya, A.; Latorre, A. Diet shapes the gut microbiota of the omnivorous cockroach blattella germanica. FEMS Microbiol. Ecol. 2015, 91, fiv022. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Z.; Ma, L.; Li, G.; Han, K.; Liu, Z.; Wang, H.; Xu, B. The native dietary habits of the two sympatric bee species and their effects on shaping midgut microorganisms. Front. Microbiol. 2021, 12, 738226. [Google Scholar] [CrossRef]
- Ptaszynska, A.A.; Latoch, P.; Hurd, P.J.; Polaszek, A.; Michalska-Madej, J.; Grochowalski, L.; Strapagiel, D.; Gnat, S.; Zaluski, D.; Gancarz, M.; et al. Amplicon sequencing of variable 16s rrna from bacteria and its2 regions from fungi and plants, reveals honeybee susceptibility to diseases results from their forage availability under anthropogenic landscapes. Pathogens 2021, 10, 381. [Google Scholar] [CrossRef]
- Hori, C.; Song, R.; Matsumoto, K.; Matsumoto, R.; Minkoff, B.B.; Oita, S.; Hara, H.; Takasuka, T.E. Proteomic characterization of lignocellulolytic enzymes secreted by the insect-associated fungus daldinia decipiens oita, isolated from a forest in northern Japan. Appl. Environ. Microbiol. 2020, 86, e02350-19. [Google Scholar] [CrossRef]
- Kirsch, R.; Wielsch, N.; Vogel, H.; Svatos, A.; Heckel, D.G.; Pauchet, Y. Combining proteomics and transcriptome sequencing to identify active plant-cell-wall-degrading enzymes in a leaf beetle. BMC Genom. 2012, 13, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.B.; Xia, K.L. An index-catalogue of chinese tettigoniodea (orthopteroidea: Grylloptera). J. Orthoptera Res. 1994, 3, 15–41. [Google Scholar]
- Zhou, Z.; Shi, F.; Zhao, L. The first mitochondrial genome for the superfamily hagloidea and implications for its systematic status in ensifera. PLoS ONE 2014, 9, e86027. [Google Scholar] [CrossRef]
- Song, H.; Amédégnato, C.; Ciglianod, M.M.; Grandcolas, L.D.; Headse, S.W.; Huang, Y.; Otteg, D.; Whiting, M.F. 300 million years of diversification: Elucidating the patterns of orthopteran evolution based on comprehensive taxon and gene sampling. Cladistics 2015, 31, 621–651. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Huang, Y.; Mao, Y.; Zhang, N.; Nie, Y.; Zhang, X.; Zhou, Y.; Mao, S. The evolutionary patterns of genome size in ensifera (insecta: Orthoptera). Front. Genet. 2021, 12, 693541. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Q.; Zhou, Z.; Wu, F.; Chen, L.; Cao, Q.; Shi, F. Gut bacterial communities across 12 ensifera (orthoptera) at different feeding habits and its prediction for the insect with contrasting feeding habits. PLoS ONE 2021, 16, e0250675. [Google Scholar] [CrossRef]
- Smith, C.C.; Srygley, R.B.; Healy, F.; Swaminath, K.; Mueller, U.G. Spatial structure of the mormon cricket gut microbiome and its predicted contribution to nutrition and immune function. Front. Microbiol. 2017, 8, 801. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.H.; Stat, M.; Bunce, M.; Simmons, L.W. The influence of diet and environment on the gut microbial community of field crickets. Ecol. Evol. 2018, 8, 4704–4720. [Google Scholar] [CrossRef]
- Rubanov, A.; Russell, K.A.; Rothman, J.A.; Nieh, J.C.; McFrederick, Q.S. Intensity of nosema ceranae infection is associated with specific honey bee gut bacteria and weakly associated with gut microbiome structure. Sci. Rep. 2019, 9, 3820. [Google Scholar] [CrossRef]
- Shelomi, M.; Lin, S.S.; Liu, L.Y. Transcriptome and microbiome of coconut rhinoceros beetle (oryctes rhinoceros) larvae. BMC Genom. 2019, 20, 957. [Google Scholar] [CrossRef] [Green Version]
- Kakumanu, M.L.; Maritz, J.M.; Carlton, J.M.; Schal, C. Overlapping community compositions of gut and fecal microbiomes in lab-reared and field-collected german cockroaches. Appl. Environ. Microbiol. 2018, 84, e01037-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.W.; Gwak, H.J.; Moon, S.; Rho, M.; Ryu, J.H. Functional dynamics of bacterial species in the mouse gut microbiome revealed by metagenomic and metatranscriptomic analyses. PLoS ONE 2020, 15, e0227886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobay, L.M.; Wissel, E.F.; Raymann, K. Strain structure and dynamics revealed by targeted deep sequencing of the honey bee gut microbiome. Msphere 2020, 5, e00694-20. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. Quast: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Hu, H.; da Costa, R.R.; Pilgaard, B.; Schiott, M.; Lange, L.; Poulsen, M. Fungiculture in termites is associated with a mycolytic gut bacterial community. Msphere 2019, 4, e00165-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, K.R.; Sanders, J.G.; Song, S.J.; Nute, M.; Metcalf, J.L.; Thompson, L.R.; Morton, J.T.; Amir, A.; McKenzie, V.J.; Humphrey, G.; et al. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 2019, 13, 576–587. [Google Scholar] [CrossRef]
- Rausch, P.; Ruhlemann, M.; Hermes, B.M.; Doms, S.; Dagan, T.; Dierking, K.; Domin, H.; Fraune, S.; von Frieling, J.; Hentschel, U.; et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 2019, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Tap, J.; Storsrud, S.; Le Neve, B.; Cotillard, A.; Pons, N.; Dore, J.; Ohman, L.; Tornblom, H.; Derrien, M.; Simren, M. Diet and gut microbiome interactions of relevance for symptoms in irritable bowel syndrome. Microbiome 2021, 9, 74. [Google Scholar] [CrossRef]
- Marynowska, M.; Goux, X.; Sillam-Dusses, D.; Rouland-Lefevre, C.; Halder, R.; Wilmes, P.; Gawron, P.; Roisin, Y.; Delfosse, P.; Calusinska, M. Compositional and functional characterisation of biomass-degrading microbial communities in guts of plant fibre- and soil-feeding higher termites. Microbiome 2020, 8, 96. [Google Scholar] [CrossRef]
- Fu, X.; Ou, Z.; Zhang, M.; Meng, Y.; Li, Y.; Wen, J.; Hu, Q.; Zhang, X.; Norback, D.; Deng, Y.; et al. Indoor bacterial, fungal and viral species and functional genes in urban and rural schools in shanxi province, china-association with asthma, rhinitis and rhinoconjunctivitis in high school students. Microbiome 2021, 9, 138. [Google Scholar] [CrossRef]
- Slizen, M.V.; Galzitskaya, O.V. Comparative analysis of proteomes of a number of nosocomial pathogens by kegg modules and kegg pathways. Int. J. Mol. Sci. 2020, 21, 7839. [Google Scholar] [CrossRef] [PubMed]
- Maire, J.; Girvan, S.K.; Barkla, S.E.; Perez-Gonzalez, A.; Suggett, D.J.; Blackall, L.L.; van Oppen, M.J.H. Intracellular bacteria are common and taxonomically diverse in cultured and in hospite algal endosymbionts of coral reefs. ISME J. 2021, 15, 2028–2042. [Google Scholar] [CrossRef] [PubMed]
- Liew, K.J.; Liang, C.H.; Lau, Y.T.; Yaakop, A.S.; Chan, K.G.; Shahar, S.; Shamsir, M.S.; Goh, K.M. Thermophiles and carbohydrate-active enzymes (cazymes) in biofilm microbial consortia that decompose lignocellulosic plant litters at high temperatures. Sci. Rep. 2022, 12, 2850. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sun, J.; Liu, C.; Yu, X.; Li, H.; Zhang, W.; Li, Y.; Geng, Y.; Wang, Z. Compositional alterations of gut microbiota in patients with diabetic kidney disease and type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 2022, 15, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Mikaelyan, A.; Dietrich, C.; Kohler, T.; Poulsen, M.; Sillam-Dusses, D.; Brune, A. Diet is the primary determinant of bacterial community structure in the guts of higher termites. Mol. Ecol. 2015, 24, 5284–5295. [Google Scholar] [CrossRef]
- Lyu, T.; Liu, G.; Zhang, H.; Wang, L.; Zhou, S.; Dou, H.; Pang, B.; Sha, W.; Zhang, H. Changes in feeding habits promoted the differentiation of the composition and function of gut microbiotas between domestic dogs (canis lupus familiaris) and gray wolves (canis lupus). AMB Express 2018, 8, 123. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food components and dietary habits: Keys for a healthy gut microbiota composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.K.; Williams, C.V.; Junge, R.E.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’Connell, T.M.; Drea, C.M. A role for gut microbiota in host niche differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef]
- Reid, N.M.; Addison, S.L.; Macdonald, L.J.; Lloyd-Jones, G. Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (prionoplus reticularis). Appl. Environ. Microbiol. 2011, 77, 7000–7006. [Google Scholar] [CrossRef] [Green Version]
- Bovo, S.; Utzeri, V.J.; Ribani, A.; Cabbri, R.; Fontanesi, L. Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci. Rep. 2020, 10, 9279. [Google Scholar] [CrossRef]
- Xia, X.; Lan, B.; Tao, X.; Lin, J.; You, M. Characterization of spodoptera litura gut bacteria and their role in feeding and growth of the host. Front. Microbiol. 2020, 11, 1492. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16s amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, A. Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 2014, 12, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Romero Victorica, M.; Soria, M.A.; Batista-Garcia, R.A.; Ceja-Navarro, J.A.; Vikram, S.; Ortiz, M.; Ontanon, O.; Ghio, S.; Martinez-Avila, L.; Quintero Garcia, O.J.; et al. Neotropical termite microbiomes as sources of novel plant cell wall degrading enzymes. Sci. Rep. 2020, 10, 3864. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Benndorf, R.; de Beer, Z.W.; Vollmers, J.; Kaster, A.K.; Beemelmanns, C.; Poulsen, M. Comparative genomics reveals prophylactic and catabolic capabilities of actinobacteria within the fungus-farming termite symbiosis. Msphere 2021, 6, e01233-20. [Google Scholar] [CrossRef]
- Schmid, J.; Heider, D.; Wendel, N.J.; Sperl, N.; Sieber, V. Bacterial glycosyltransferases: Challenges and opportunities of a highly diverse enzyme class toward tailoring natural products. Front. Microbiol. 2016, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhou, Y.; Qu, M.; Qiu, Y.; Guo, X.; Zhang, Y.; Liu, T.; Yang, J.; Yang, Q. Structural and biochemical insights into the catalytic mechanisms of two insect chitin deacetylases of the carbohydrate esterase 4 family. J. Biol. Chem. 2019, 294, 5774–5783. [Google Scholar] [CrossRef] [Green Version]
- Benjamino, J.; Lincoln, S.; Srivastava, R.; Graf, J. Low-abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome 2018, 6, 86. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomy | Feeding Habits | Location | Abbreviations | ||
|---|---|---|---|---|---|
| Family | Species | County/Mountain, Province | Geographic Coordinates | ||
| Tettigoniidae | Mecopoda niponensis | Herbivore | Tianmu Mountains National Nature Reserve, Zhejiang | 30°35′ N 119°43′ E | Mec |
| Gryllacrididae | Ocellarnaca emeiensis | Carnivore | Daming Mountains National Nature Reserve, Guangxi | 23°52′ N 108°34′ E | Oce |
| Gryllotalpidae | Gryllotalpa orientalis | Omnivore | Quyang County, Hebei | 38°59′ N 114°78′ E | Gry |
| Samples | Gryllotalpa orientalis | Mecopoda niponensis | Ocellarnaca emeiensis | ||||
|---|---|---|---|---|---|---|---|
| Microbial Taxonomy | Phylum/% | Species/% | Phylum/% | Species/% | Phylum/% | Species/% | |
| Identification of microbiota | Top1 | Firmicutes/47.09 | Intestinimonas massiliensis/42.12 | Proteobacteria/77.65 | Leclercia adecarboxy lata/12.80 | Proteobacteria/40.34 | Lactococcus lactis/23.82 |
| Top2 | Proteobacteria/31.80 | Acinetobacter baumannii/15.42 | Mucoromycota/7.43 | Kluyvera cryocrescens/9.51 | Firmicutes/20.04 | Acinetobacter baumannii/20.86 | |
| Top3 | Bacteroidetes/8.69 | Cotesia sesamiae bracovirus/10.82 | Basidiomycota/2.94 | Pseudomonas aeruginosa/9.16 | Basidiomycota/14.64 | Rhizophagus irregularis/8.16 | |
| Top4 | Candidatus_Tectomicrobia/2.61 | Pseudomonas aeruginosa/7.8 | Firmicutes/2.51 | Acinetobacter baumannii/8.47 | Mucoromycota/7.01 | Solemya velum gill symbiont/8.14 | |
| KEGG Pathway | Percent of Genes (%) | ||
|---|---|---|---|
| Mec | Oce | Gry | |
| Organismal systems | 0.00 | 0.06 | 0.00 |
| Metabolism | 72.78 | 65.35 | 10.95 |
| Human diseases | 1.86 | 2.03 | 83.60 |
| Genetic information processing | 8.66 | 17.58 | 4.00 |
| Environmental information processing | 11.22 | 10.49 | 0.79 |
| Cellular processes | 5.48 | 4.49 | 0.66 |
| Groups | High Abundance CAZymes | Significantly Different CAZymes | Unique CAZymes | |||
|---|---|---|---|---|---|---|
| CAZyme Family | Activities in Family | CAZyme Family | Activities in family | CAZyme Family | Activities in Family | |
| Mec | GT47 | heparan β-glucuronyltransferase (EC 2.4.1.225); xyloglucan β-galactosyltransferase (EC 2.4.1.-) | CE3 | acetyl xylan esterase (EC 3.1.1.72) | CE3 | acetyl xylan esterase (EC 3.1.1.72) |
| GT2 | cellulose synthase (EC 2.4.1.12); chitin synthase (EC 2.4.1.16) | CBM10 | cellulose-binding function | CBM10, | cellulose-binding function | |
| GT4 | sucrose synthase (EC 2.4.1.13); sucrose-phosphate synthase (EC 2.4.1.14) | GH5_18 | b-mannosidase (EC 3.2.1.25) | |||
| GH5_13 | b-D-galactofuranosidase (EC 3.2.1.146);a-L-arabinofuranosidase (EC 3.2.1.55) | |||||
| GH100 | alkaline and neutral invertase (EC 3.2.1.26) | |||||
| Oce | GT47 | heparan β-glucuronyltransferase (EC 2.4.1.225); xyloglucan β-galactosyltransferase (EC 2.4.1.-) | CE8 | pectin methylesterase (EC 3.1.1.11) | ||
| CE8 | pectin methylesterase (EC 3.1.1.11) | GT1 | UDP-glucuronosyltransferase (EC 2.4.1.17); zeatin O-β-xylosyltransferase (EC 2.4.2.40) | |||
| GT1 | UDP-glucuronosyltransferase (EC 2.4.1.17); zeatin O-β-xylosyltransferase (EC 2.4.2.40) | GH38 | α-mannosidase (EC 3.2.1.24); mannosyl-oligosaccharide α-1,2-mannosidase (EC 3.2.1.113) | |||
| GH17 | glucan endo-1,3-β-glucosidase (EC 3.2.1.39); licheninase (EC 3.2.1.73) | |||||
| Gry | GT2 | cellulose synthase (EC 2.4.1.12); chitin synthase (EC 2.4.1.16) | ||||
| GH3 | β-glucosidase (EC 3.2.1.21); xylan 1,4-β-xylosidase (EC 3.2.1.37) | |||||
| GT47 | heparan β-glucuronyltransferase (EC 2.4.1.225); xyloglucan β-galactosyltransferase (EC 2.4.1.-) | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Zhu, Q.; Qin, M.; Zhou, Z.; Liu, C.; Wang, L.; Shi, F. The Role of Feeding Characteristics in Shaping Gut Microbiota Composition and Function of Ensifera (Orthoptera). Insects 2022, 13, 719. https://doi.org/10.3390/insects13080719
Zheng X, Zhu Q, Qin M, Zhou Z, Liu C, Wang L, Shi F. The Role of Feeding Characteristics in Shaping Gut Microbiota Composition and Function of Ensifera (Orthoptera). Insects. 2022; 13(8):719. https://doi.org/10.3390/insects13080719
Chicago/Turabian StyleZheng, Xiang, Qidi Zhu, Meng Qin, Zhijun Zhou, Chunmao Liu, Liyuan Wang, and Fuming Shi. 2022. "The Role of Feeding Characteristics in Shaping Gut Microbiota Composition and Function of Ensifera (Orthoptera)" Insects 13, no. 8: 719. https://doi.org/10.3390/insects13080719