
A Comparative Analysis of Mitogenomes in Species of the Tapinoma nigerrimum Complex and Other Species of the Genus Tapinoma (Formicidae, Dolichoderinae)

 , , , and
, , , and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Determination of Species Identity of Focal Samples
2.2. DNA Extraction, Mitogenomic Sequencing, and Assembly Strategies
2.3. Mitogenome Annotation and Sequence Analysis
2.4. Comparative Phylogenetics
3. Results and Discussion
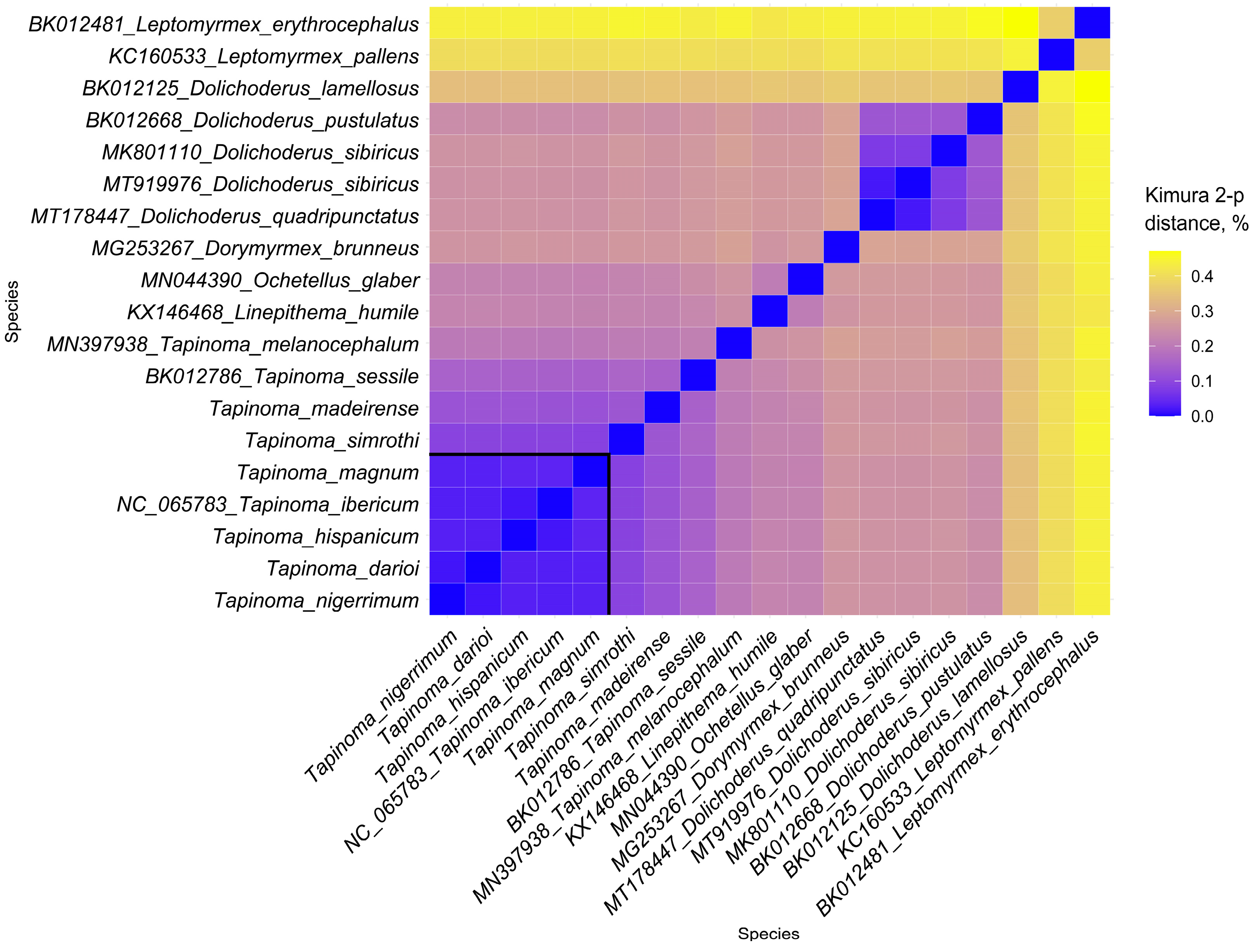
3.1. General Features and Phylogenetic Analysis
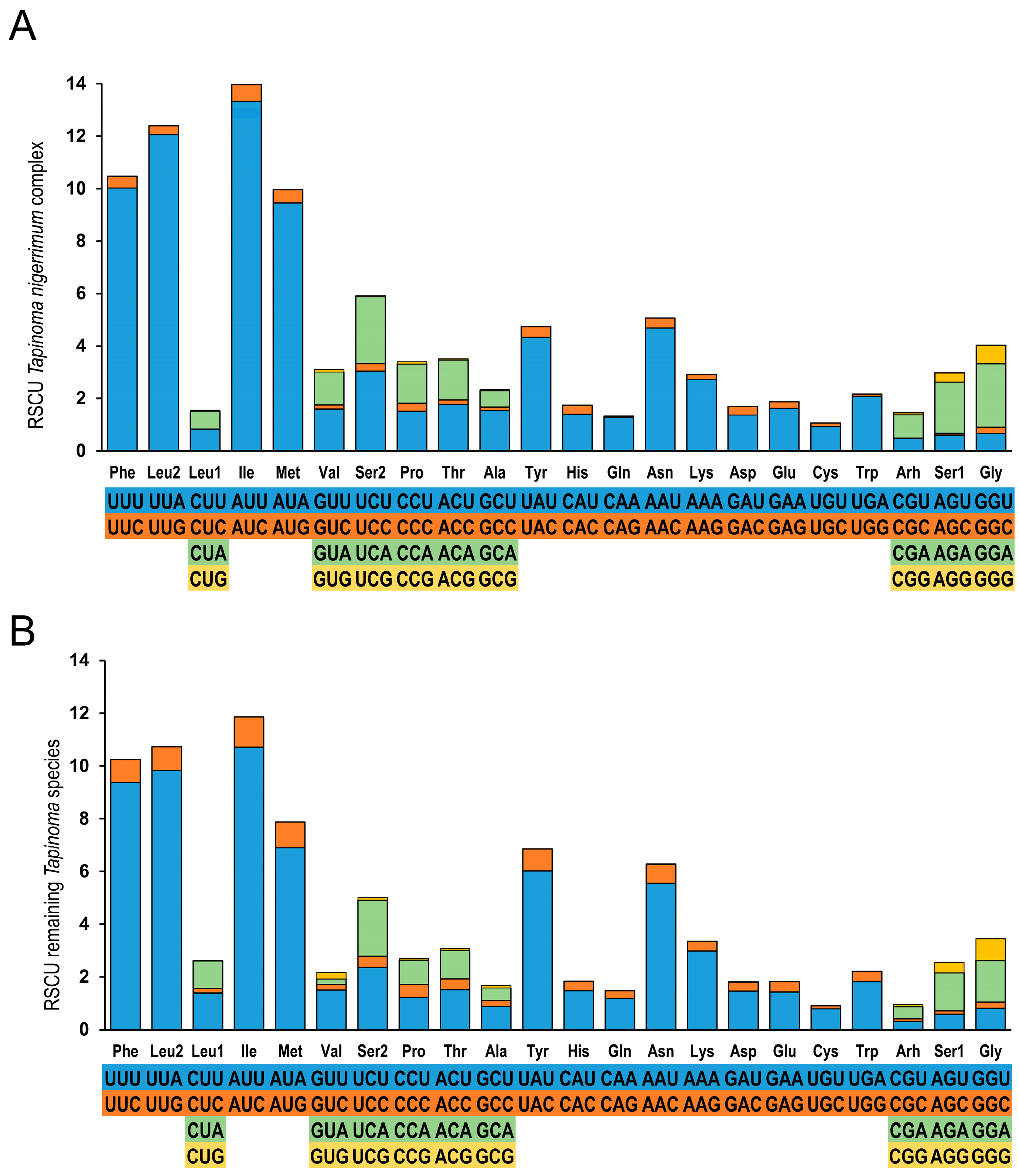
3.2. Gene Organization and Sequence Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hölldobler, B.; Wilson, E.O. The Ants; Harvard University Press: Cambridge, MA, USA, 1990. [Google Scholar]
- Lach, L.; Parr, C.; Abbott, K. Ant Ecology; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Bolton, B. An Online Catalog of the Ants of the World. Available online: http://antcat.org (accessed on 15 April 2024).
- Schultz, T.R. In search of ant ancestors. Proc. Natl. Acad. Sci. USA 2000, 97, 14028–14029. [Google Scholar] [CrossRef] [PubMed]
- Bertelsmeier, C.; Luque, G.M.; Hoffmann, B.D.; Courchamp, F. Worldwide ant invasions under climate change. Biodiver. Conserv. 2015, 24, 117–128. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Bamisile, B.S.; Khan, M.M.; Islam, W.; Hafeez, M.; Bodlah, I.; Xu, Y. Impact of invasive ant species on native fauna across similar habitats under global environmental changes. Environ. Sci. Pollut. Res. 2021, 28, 54362–54382. [Google Scholar] [CrossRef] [PubMed]
- Klimeš, P.; Okrouhlík, J. Invasive ant Tapinoma melanocephalum (Hymenoptera: Formicidae): A rare guest or increasingly common indoor pest in Europe? Eur. J. Entomol. 2015, 112, 705–712. [Google Scholar] [CrossRef]
- Smith, M.A.; Fisher, B.L. Invasions, DNA barcodes, and rapid biodiversity assessment using ants of Mauritius. Front. Zool. 2009, 6, 31. [Google Scholar] [CrossRef]
- Seifert, B.; D’Eustacchio, D.; Kaufmann, B.; Centorame, M.; Lorite, P.; Modica, M.V. Four species within the supercolonial ants of the Tapinoma nigerrimum complex revealed by integrative taxonomy (Hymenoptera: Formicidae). Myrmecol. News 2017, 24, 123–144. [Google Scholar] [CrossRef]
- Seifert, B.; Kaufmann, B.; Fraysse, L. A taxonomic revision of the Palaearctic species of the ant genus Tapinoma Mayr 1861 (Hymenoptera: Formicidae). Zootaxa 2024, 5435, 1–74. [Google Scholar] [CrossRef]
- Centanni, J.; Kaufmann, B.; Blatrix, R.; Blight, O.; Dumet, A.; Jay-Robert, P.; Vergnes, A. High resolution mapping in Southern France reveals that distributions of supercolonial and monodomous species in the Tapinoma nigerrimum complex (Hymenoptera: Formicidae) are related to sensitivity to urbanization. Myrmecol. News 2022, 32, 41–50. [Google Scholar] [CrossRef]
- Flucher, S.M.; Krapf, P.; Arthofer, W.; Suarez, A.V.; Crozier, R.H.; Steiner, F.M.; Schlick-Steiner, B.C. Effect of social structure and introduction history on genetic diversity and differentiation. Mol. Ecol. 2021, 30, 2511–2527. [Google Scholar] [CrossRef]
- Blight, O.; Provost, E.; Renucci, M.; Tirard, A.; Orgeas, J. A native ant armed to limit the spread of the Argentine ant. Biol. Invasions 2010, 12, 3785–3793. [Google Scholar] [CrossRef]
- Zina, V.; Conde, S.; Branco, M.; Franco, J.C. Do dominant native ants outcompete the invasive Argentine ant in Mediterranean citrus ecosystems? A laboratory test. Insects 2024, 15, 333. [Google Scholar] [CrossRef] [PubMed]
- Seifert, B. Hybridization in the European carpenter ants Camponotus herculeanus and C. ligniperda (Hymenoptera: Formicidae). Insect. Soc. 2019, 66, 365–374. [Google Scholar] [CrossRef]
- Ruiz-Mena, A.; Mora, P.; Montiel, E.E.; Palomeque, T.; Lorite, P. Complete nucleotide sequence of the mitogenome of Tapinoma ibericum (Hymenoptera: Formicidae: Dolichoderinae), gene organization and phylogenetic implications for the Dolichoderinae subfamily. Genes 2022, 13, 1325. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hu, S.Y.; Li, M.; Sun, H.; Yuan, M.L. Comparative mitogenomic analysis provides evolutionary insights into Formica (Hymenoptera: Formicidae). PLoS ONE 2024, 19, e0302371. [Google Scholar] [CrossRef] [PubMed]
- Seifert, B.; Kulmuni, J.; Pamilo, P. Independent hybrid populations of Formica polyctena X rufa wood ants (Hymenoptera: Formicidae) abound under conditions of forest fragmentation. Evol. Ecol. 2010, 24, 1219–1237. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: A decade of progress. Annu. Rev. Entomol, 2024; advance online publication. [Google Scholar] [CrossRef]
- Bernt, M.; Bleidorn, C.; Braband, A.; Dambach, J.; Donath, A.; Fritzsch, G.; Golombek, A.; Hadrys, H.; Jühling, F.; Meusemann, K.; et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Phylogenetics Evol. 2013, 69, 352–364. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Cameron, S.L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef]
- Park, J.; Xi, H.; Park, J. Comparative analysis on Palaearctic Dolichoderus quadripunctatus species group with the proposal of a novel species (Formicidae). In Proceedings of the 2020 Spring International Conference of Korean Society of Applied Entomology, Online Academic Presentation (Society’s Website), 25–29 May 2020. [Google Scholar] [CrossRef]
- Park, J.; Kwon, W.; Park, J. The complete mitochondrial genome of Siberian odorous ant, Dolichoderus sibiricus Emery, 1889 (Hymenoptera: Formicidae). Mitochondrial DNA Part B 2019, 4, 525–526. [Google Scholar] [CrossRef]
- Berman, M.; Austin, C.M.; Miller, A.D. Characterisation of the complete mitochondrial genome and 13 microsatellite loci through next-generation sequencing for the New Caledonian spider-ant Leptomyrmex pallens. Mol. Biol. Rep. 2014, 41, 1179–1187. [Google Scholar] [CrossRef]
- Zhao, E.; Bi, G.; Yang, J.; Zhang, Z.; Liu, G.; Du, Q.; Shang, E. Complete mitochondrial genome of the Argentine ant, Linepithema humile (Hymenoptera: Formicidae). Mitochondrial DNA Part A 2017, 28, 210–211. [Google Scholar] [CrossRef]
- Duan, X.Y.; Peng, X.Y.; Qian, Z.Q. The complete mitochondrial genomes of two globally invasive ants, the Argentine ant Linepithema humile and the little fire ant Wasmannia auropunctata. Conserv. Genet. Resour. 2016, 8, 275–277. [Google Scholar] [CrossRef]
- Park, J.; Park, C.H.; Park, J. Complete mitochondrial genome of the H3 haplotype Argentine ant Linepithema humile (Mayr, 1868) (Formicidae; Hymenoptera). Mitochondrial DNA Part B 2021, 6, 786–788. [Google Scholar] [CrossRef]
- Park, J.; Xi, H.; Park, J. The complete mitochondrial genome of Ochetellus glaber (Mayr, 1862) (Hymenoptera: Formicidae). Mitochondrial DNA Part B 2020, 5, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Song, X.; Yu, H.; Lu, Z. Complete mitochondrial genome sequence of Tapinoma melanocephalum (Hymenoptera: Formicidae). Mitochondrial DNA Part B 2019, 4, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Borowiec, M.L.; Zhang, Y.M.; Neves, K.; Ramalho, M.O.; Fisher, B.L.; Lucky, A.; Moreau, C.S. Evaluating UCE data adequacy and integrating uncertainty in a comprehensive phylogeny of ants. bioRxiv 2024. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Ward, P.S.; Brady, S.G.; Fisher, B.L.; Schultz, T.R. Phylogeny and biogeography of dolichoderine ants: Effects of data partitioning and relict taxa on historical inference. Syst. Biol. 2010, 59, 342–362. [Google Scholar] [CrossRef]
- Moreau, C.S.; Bell, C.D. Testing the museum versus cradle tropical biological diversity hypothesis: Phylogeny, diversification, and ancestral biogeographic range evolution of the ants. Evolution 2013, 67, 2240–2257. [Google Scholar] [CrossRef]
- Seifert, B. The Gene and Gene Expression (GAGE) species concept: An universal approach for all eukaryotic organisms. Syst. Biol. 2020, 69, 1033–1038. [Google Scholar] [CrossRef]
- Seifert, B. Frequent misclassification by mtDNA barcoding as revealed by nuDNA and/or testable analysis of its expression products. Biodivers. J. 2024, 15, 129–133. [Google Scholar] [CrossRef]
- Boore, J.; Lavrov, D.; Brown, W. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Sterling-Montealegre, R.A.; Prada, C.F. Variability and evolution of gene order rearrangement in mitochondrial genomes of arthropods (except Hexapoda). Gene 2023, 892, 147906. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Tyagi, K.; Dubey, A.K. Purifying selection drove the adaptation of mitochondrial genes along with correlation of gene rearrangements and evolutionary rates in two subfamilies of whitefly (Insecta: Hemiptera). Funct. Integr. Genom. 2024, 24, 121. [Google Scholar] [CrossRef] [PubMed]
- Gotzek, D.; Clarke, J.; Shoemaker, D. Mitochondrial genome evolution in fire ants (Hymenoptera: Formicidae). BMC Evol. Biol. 2010, 10, 300. [Google Scholar] [CrossRef]
- Rodovalho, C.d.M.; Lyra, M.L.; Ferro, M.; Bacci, M., Jr. The mitochondrial genome of the leaf-cutter ant Atta laevigata: A mitogenome with a large number of intergenic spacers. PLoS ONE 2014, 9, e97117. [Google Scholar] [CrossRef]
- Beckenbach, A.T.; Stewart, J.B. Insect mitochondrial genomics 3: The complete mitochondrial genome sequences of representatives from two neuropteroid orders: A dobsonfly (order Megaloptera) and a giant lacewing and an owlfly (order Neuroptera). Genome 2009, 52, 31–38. [Google Scholar] [CrossRef]
- Wei, S.J.; Tang, P.; Zheng, L.H.; Shi, M.; Chen, X.X. The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A+T content and a long intergenic spacer between atp8 and atp6. Mol. Biol. Rep. 2010, 37, 1931–1942. [Google Scholar] [CrossRef]
- Zheng, B.Y.; Cao, L.J.; Tang, P.; van Achterberg, K.; Hoffmann, A.A.; Chen, H.Y.; Chen, X.X.; Wei, S.J. Gene arrangement and sequence of mitochondrial genomes yield insights into the phylogeny and evolution of bees and sphecid wasps (Hymenoptera: Apoidea). Mol. Phylogenetics Evol. 2018, 124, 1–9. [Google Scholar] [CrossRef]
- Sun, Z.; Wan, D.G.; Murphy, R.W.; Ma, L.; Zhang, X.S.; Huang, D.W. Comparison of base composition and codon usage in insect mitochondrial genomes. Genes Genom. 2009, 31, 65–71. [Google Scholar] [CrossRef]
- Pita, S.; Panzera, F.; Vela, J.; Mora, P.; Palomeque, T.; Lorite, P. Complete mitochondrial genome of Triatoma infestans (Hemiptera, Reduviidae, Triatominae), main vector of Chagas disease. Infect. Genet. Evol. 2017, 54, 158–163. [Google Scholar] [CrossRef]
- Singh, D.; Kabiraj, D.; Sharma, P.; Chetia, H.; Mosahari, P.V.; Neog, K.; Bora, U. The mitochondrial genome of Muga silkworm (Antheraea assamensis) and its comparative analysis with other lepidopteran insects. PLoS ONE 2017, 12, e0188077. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.K.; Chen, Q.P.; Ayivi, S.P.G.; Guan, J.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Three complete mitochondrial genomes of Orestes guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis (Insecta: Phasmatodea) and their phylogeny. Insects 2021, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; He, D.; Guang, X.; Li, Q. Novel tRNA gene rearrangements in the mitochondrial genomes of Poneroid ants and phylogenetic implication of Paraponerinae (Hymenoptera: Formicidae). Life 2023, 13, 2068. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Pons, J.; Bover, P.; Bidegaray-Batista, L.; Arnedo, M.A. Armless mitochondrial tRNAs conserved for over 30 million years in spiders. BMC Genom. 2019, 20, 665. [Google Scholar] [CrossRef]
- Lin, X.; Song, N. The first complete mitochondrial genome of the genus Pachycondyla (Formicidae, Ponerinae) and insights into the phylogeny of ants. Genes 2023, 14, 1528. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, Q.; Li, M.; Wei, J.; Zhang, X.; Zhang, H. Comparative mitogenomic analysis of the Eurydema genus in the context of representative Pentatomidae (Hemiptera: Heteroptera) taxa. J. Insect Sci. 2019, 19, 20. [Google Scholar] [CrossRef]
- Li, K.; Liang, A.-P. Hemiptera mitochondrial control region: New sights into the structural organization, phylogenetic utility, and roles of tandem repetitions of the noncoding segment. Int. J. Mol. Sci. 2018, 19, 1292. [Google Scholar] [CrossRef]
- Zhang, X.M.; Li, T.; Liu, X.; Xu, Z.H. Characterization and phylogenetic implication of complete mitochondrial genome of the medicinal ant Formica sinae (Hymenoptera: Formicidae): Genomic comparisons in Formicidae. J. Med. Entomol. 2022, 59, 1971–1979. [Google Scholar] [CrossRef]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | Coordinates | Locality Nest | Collection Date |
|---|---|---|---|
| T. darioi | 40.65401° N, 0.411551° E | Benicarló (Spain) | 20 September 2019 |
| T. magnum | 46.20167° N, 4.828° E | Lyon (France) | 2011 |
| T. hispanicum | 37.7718° N, 3.4972° W | Torres (Spain) | 31 October 2014 |
| T. nigerrimum | 43.684254° N, 3.876259° E | Prades-le-Lez (France) | 30 April 2012 |
| T. simrothi | 37.714281° N, 3.904023° W | Jamilena (Spain) | 23 April 2014 |
| T. madeirense | 37.259167° N, 3.487777° W | Sierra de Huetor (Spain) | 19 April 2004 |
| Species | Mitogenome Size (bp) | Country of Origin | Accession Number | Reference |
|---|---|---|---|---|
| Tribe Dolichoderini | ||||
| Dolichoderus lamellosus (Mayr, 1870) | 16,234 | Costa Rica | BK012125 | [30] |
| Dolichoderus pustulatus (Mayr, 1886) | 16,224 | Canada | BK012668 | [30] |
| Dolichoderus quadripunctatus (Linnaeus, 1771) | 16,017 | Poland | MT178447 | [31] |
| Dolichoderus sibiricus (Emery, 1889) | 16,086 | South Korea | MH719017 | [32] |
| 16,044 | South Korea | MK801110 | [31] | |
| 16,067 | Russia | MT919976 | unpublished | |
| 16,110 | Taiwan | MW160468 | unpublished | |
| Tribe Leptomyrmecini | ||||
| Dorymyrmex brunneus (Forel, 1908) | 15,848 | - | MG253267 | unpublished |
| Leptomyrmex erythrocephalus (Fabricius, 1775) | 15,546 | Australia | BK012481 | [30] |
| Leptomyrmex pallens (Emery, 1883) | 15,591 | New Calcedonia | KC160533 | [33] |
| Linepithema humile (Mayr, 1868) | 16,098 | USA | KT428891 | [34] |
| 15,929 | - | KX146468 | [35] | |
| 15,934 | South Korea | MT890564 | [36] | |
| Ochetellus glaber (Mayr, 1862) | 16,259 | South Korea | MN044390 | [37] |
| Tribe Tapinomini | ||||
| Tapinoma darioi (Seifert et al., 2017) | 15,680 | Spain | PQ459328 | This study |
| Tapinoma hispanicum (Seifert et al., 2024) | 15,665 | Spain | PQ459329 | This study |
| Tapinoma ibericum (Santschi, 1925) | 15,715 | Spain | NC_065783 | [16] |
| Tapinoma madeirense (Forel, 1895) | 15,507 | Spain | PQ459330 | This study |
| Tapinoma magnum (Mayr, 1861) | 15,694 | France | PQ459331 | This study |
| Tapinoma melanocephalum (Fabricius, 1793) | 15,499 | China | MN397938 | [38] |
| Tapinoma nigerrimum (Nylander, 1856) | 15,817 | France | PQ459332 | This study |
| Tapinoma sessile (Say, 1836) | 15,287 | USA | BK012786 | [30] |
| Tapinoma simrothi (Krausse, 1911) | 15,487 | Spain | PQ459333 | This study |
| Start codons | |||||||||||||
| nad2 | cox1 | cox2 | atp8 | atp6 | cox3 | nad3 | nad5 | nad4 | nad4l | nad6 | cob | nad1 | |
| T. nigerrimum | ATA | ATG | ATT | ATC | ATG | ATG | ATA | ATA | ATG | ATT | ATG | ATG | ATT |
| T. darioi | ATA | ATG | ATT | ATC | ATG | ATG | ATA | ATA | ATG | ATT | ATG | ATG | ATT |
| T. hispanicum | ATA | ATG | ATT | ATC | ATG | ATG | ATA | ATA | ATG | ATT | ATG | ATG | ATT |
| T. ibericum | ATA | ATG | ATT | ATC | ATG | ATG | ATA | ATA | ATG | ATT | ATG | ATG | ATT |
| T. magnum | ATA | ATG | ATT | ATT | ATG | ATG | ATA | ATA | ATG | ATT | ATG | ATG | ATT |
| T. simrothi | ATA | ATG | ATT | ATA | ATG | ATG | ATA | ATA | ATG | ATT | ATG | ATG | ATT |
| T. madeirense | ATA | ATG | ATT | ATA | ATG | ATG | ATT | ATT | ATG | ATT | ATG | ATG | ATA |
| T. sessile | ATA | ATG | ATT | ATA | ATG | ATG | ATT | ATT | ATG | ATT | ATG | ATG | ATT |
| T. melanocephalum | ATA | ATG | ATT | ATT | ATG | ATG | ATA | ATA | ATG | ATT | ATT | ATG | ATT |
| Stop codons | |||||||||||||
| nad2 | cox1 | cox2 | atp8 | atp6 | cox3 | nad3 | nad5 | nad4 | nad4l | nad6 | cob | nad1 | |
| T. nigerrimum | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA |
| T. darioi | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA |
| T. hispanicum | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA |
| T. ibericum | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA |
| T. magnum | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA |
| T. simrothi | TA- | TA- | TAA | TAA | TAA | TAA | TAA | TA- | TAA | TAA | TAA | TAA | TAA |
| T. madeirense | TAA | TA- | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA | TAA |
| T. sessile | TAA | TA- | TAA | TAA | TAA | TAA | TAA | TA- | TAA | TAA | TAA | TAA | TAA |
| T. melanocephalum | T-- | TA- | TAA | TAA | TAA | TAA | TAA | T- | TAA | TAA | TAA | TAA | TAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Mena, A.; Mora, P.; Rico-Porras, J.M.; Kaufmann, B.; Seifert, B.; Palomeque, T.; Lorite, P. A Comparative Analysis of Mitogenomes in Species of the Tapinoma nigerrimum Complex and Other Species of the Genus Tapinoma (Formicidae, Dolichoderinae). Insects 2024, 15, 957. https://doi.org/10.3390/insects15120957
Ruiz-Mena A, Mora P, Rico-Porras JM, Kaufmann B, Seifert B, Palomeque T, Lorite P. A Comparative Analysis of Mitogenomes in Species of the Tapinoma nigerrimum Complex and Other Species of the Genus Tapinoma (Formicidae, Dolichoderinae). Insects. 2024; 15(12):957. https://doi.org/10.3390/insects15120957
Chicago/Turabian StyleRuiz-Mena, Areli, Pablo Mora, José M. Rico-Porras, Bernard Kaufmann, Bernhard Seifert, Teresa Palomeque, and Pedro Lorite. 2024. "A Comparative Analysis of Mitogenomes in Species of the Tapinoma nigerrimum Complex and Other Species of the Genus Tapinoma (Formicidae, Dolichoderinae)" Insects 15, no. 12: 957. https://doi.org/10.3390/insects15120957
APA StyleRuiz-Mena, A., Mora, P., Rico-Porras, J. M., Kaufmann, B., Seifert, B., Palomeque, T., & Lorite, P. (2024). A Comparative Analysis of Mitogenomes in Species of the Tapinoma nigerrimum Complex and Other Species of the Genus Tapinoma (Formicidae, Dolichoderinae). Insects, 15(12), 957. https://doi.org/10.3390/insects15120957