
The Fast Evolution of the Stenobothrini Grasshoppers (Orthoptera, Acrididae, and Gomphocerinae) Revealed by an Analysis of the Control Region of mtDNA, with an Emphasis on the Stenobothrus eurasius Group

Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Sample Collection and DNA Extraction
2.2. DNA Sequencing
2.3. Sliding Window Analysis
2.4. Phylogenetic Analysis
2.5. Distribution Map
3. Results
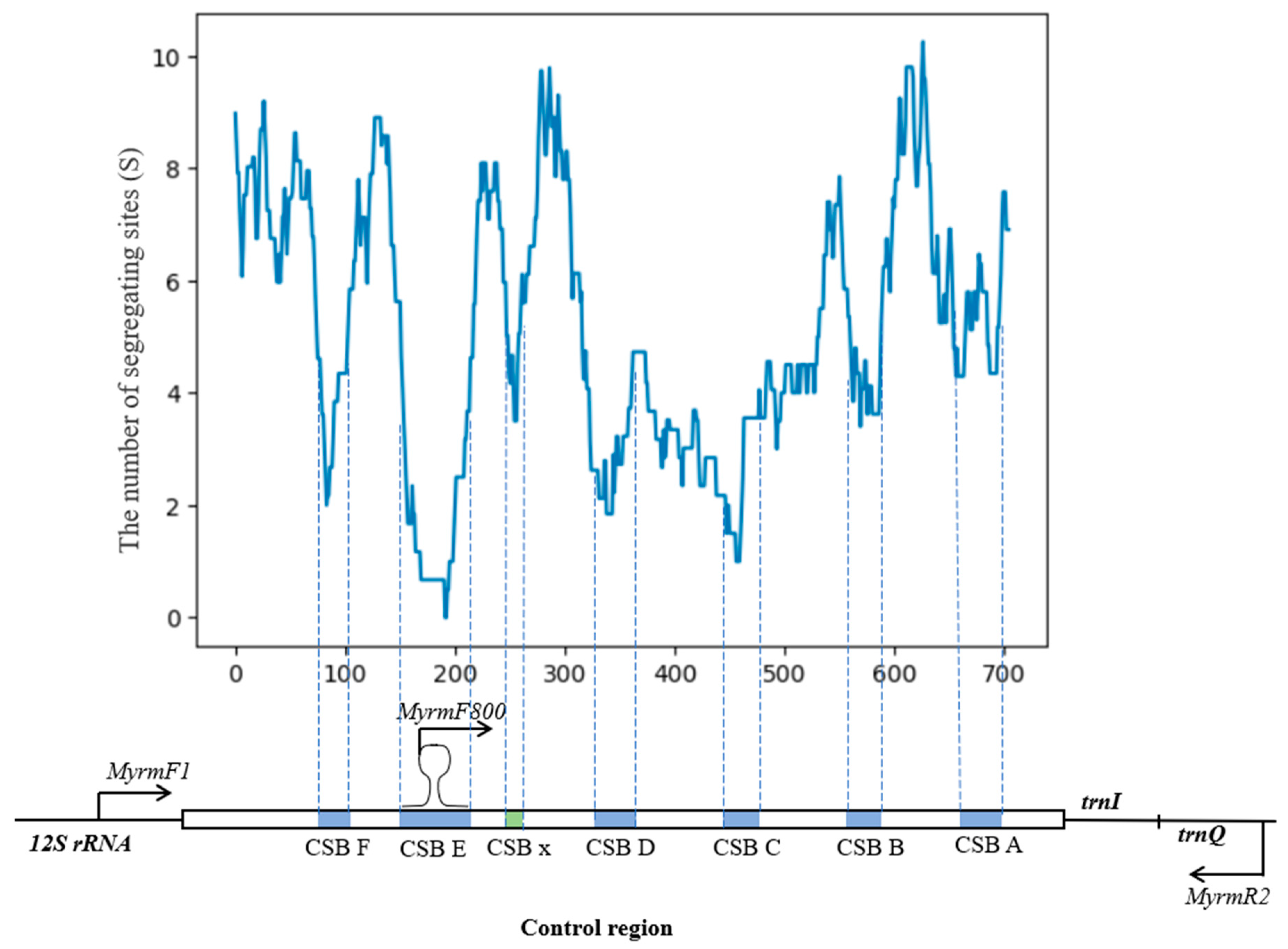
3.1. The Control Region Structure
3.2. Phylogenetic Reconstructions and Substitution Rates of the Control Region and the COI Gene
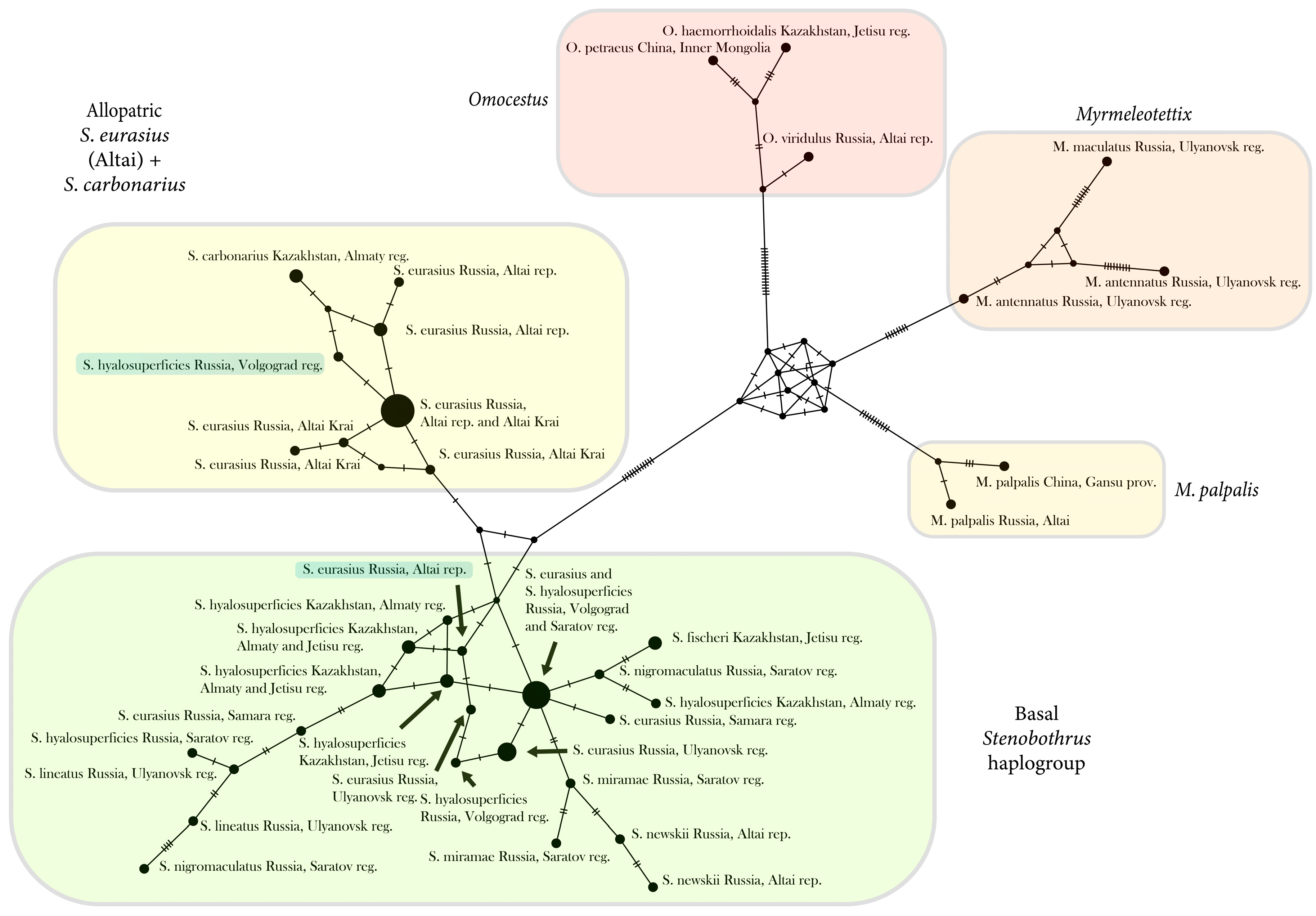
3.3. The Network Based on the Control Region
4. Discussion
4.1. Structure and Evolutionary Rate of the mtDNA Control Region
4.2. Phylogenetic Relationships and Taxonomy within the Tribe Stenobothrini
4.3. Phylogeography of the Genus Stenobothrus and Possible Scenarios of the Origin of S. eurasius and S. hyalosuperficies
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobzhansky, T. Genetics and the Origin of Species, 1st ed.; Columbia University Press: New York, NY, USA, 1937. [Google Scholar]
- Mayr, E. Populations, Species, and Evolution; Belknap Press: Cambridge, MA, USA, 1970; 453p. [Google Scholar]
- Coyne, J.A.; Orr, H.A. A catalogue and critique of species concepts. In Speciation; Oxford University Press: Oxford, UK, 2004; pp. 272–292. [Google Scholar]
- West–Eberhard, M.J. Sexual selection, social competition, and speciation. Q. Rev. Biol. 1983, 58, 155–183. [Google Scholar] [CrossRef]
- Andersson, M. Sexual Selection; Princeton University Press: Princeton, NJ, USA, 1994; Volume 72. [Google Scholar]
- Panhuis, T.M.; Butlin, R.; Zuk, M.; Tregenza, T. Sexual selection and speciation. Trends Ecol. Evol. 2001, 16, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Ragge, D.R.; Reynolds, W.J. The Songs of the Grasshoppers and Crickets of Western Europe; Harley Books, Natural History Museum: London, UK, 1998; 591p. [Google Scholar]
- Bukhvalova, M.A. Acoustic signals and morphological features of some grasshoppers of the Chorthippus biguttulus group (Orthoptera, Acrididae) of Russia and adjacent territories. Entomol. Rev. 1995, 74, 56–67. [Google Scholar]
- Vedenina, V.Y.; von Helversen, O. A re–examination of the taxonomy of the Chorthippus albomarginatus group in Europe on the basis of song and morphology (Orthoptera: Acrididae). Tijdschr. Voor Entomol. 2009, 152, 65–97. [Google Scholar] [CrossRef]
- Tarasova, T.A.; Sevastianov, N.S.; Vedenina, V.Y. Songs and morphology in grasshoppers of the Stenobothrus eurasius group (Orthoptera: Acrdidae: Gomphocerinae) from Russia and adjacent countries: Clarifying of taxonomic status. Zootaxa 2021, 4965, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Vedenina, V.; Mugue, N. Speciation in gomphocerine grasshoppers: Molecular phylogeny versus bioacoustics and courtship behavior. J. Orthoptera Res. 2011, 20, 109–125. [Google Scholar] [CrossRef]
- Vedenina, V.Y.; Sevastianov, N.S.; Tarasova, T.A. Contributions to the study of the grasshopper (Orthoptera: Acrididae: Gomphocerinae) courtship songs from Kazakhstan and adjacent territories. Zootaxa 2020, 4895, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Bridle, J.R.; Butlin, R.K. Mating signal variation and bimodality in a mosaic hybrid zone between Chorthippus grasshopper species. Evolution. 2002, 56, 1184–1198. [Google Scholar] [CrossRef]
- Vedenina, V.Y.; von Helversen, O. Complex courtship in a bimodal grasshopper hybrid zone. Behav. Ecol. Sociobiol. 2003, 54, 44–54. [Google Scholar] [CrossRef]
- Vedenina, V.Y.; Sradnick, J.; Klopfel, A.; Elsner, N. A narrow hybrid zone between the grasshoppers Stenobothrus clavatus and Stenobothrus rubicundus: Courtship song analysis. Biol. J. Linn. Soc. 2012, 107, 383–397. [Google Scholar] [CrossRef]
- Tarasova, T.; Sevastianov, N.; Vedenina, V. The processes in the hybrid zone between sibling grasshopper species of the Stenobothrus eurasius group (Acrididae, Gomphocerinae). In Proceedings of the Invertebrate Sound and Vibration Conference, Lincoln, UK, 30 March–2 April 2023; p. 29. [Google Scholar]
- Sevastianov, N.; Neretina, T.; Vedenina, V. Evolution of calling songs in the grasshopper subfamily Gomphocerinae (Orthoptera, Acrididae). Zool. Scr. 2023, 52, 154–175. [Google Scholar] [CrossRef]
- Hawlitschek, O.; Ortiz, E.M.; Noori, S.; Webster, K.C.; Husemann, M.; Pereira, R.J. Transcriptomic data reveals nuclear-mitochondrial discordance in Gomphocerinae grasshoppers (Insecta: Orthoptera: Acrididae). Mol. Phylogen. Evol. 2022, 170, 107439. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Hill, J.G.; Ortego, J.; Knowles, L.L. Paraphyletic species no more –genomic data resolve a Pleistocene radiation and validate morphological species of the Melanoplus scudderi complex (Insecta: Orthoptera). Syst. Entomol. 2020, 45, 594–605. [Google Scholar] [CrossRef]
- Hawlitschek, O.; Morinière, J.; Lehmann, G.U.C.; Lehmann, A.W.; Kropf, M.; Dunz, A.; Glaw, F.; Detcharoen, M.; Schmidt, S.; Hausmann, A.; et al. DNA barcoding of crickets, katydids and grasshoppers (Orthoptera) from Central Europe with focus on Austria, Germany and Switzerland. Mol. Ecol. Res. 2017, 17, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Gaugel, S.M.; Hawlitschek, O.; Dey, L.S.; Husemann, M. Evolution of mitogenomic gene order in Orthoptera. Insect Mol. Biol. 2023, 32, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Mao, B.; Storozhenko, S.Y.; Huang, Y.; Chen, Z.; Huang, J. Phylogenetic position of the genus Alulacris (Orthoptera: Acrididae: Melanoplinae: Podismini) revealed by complete mitogenome evidence. Insects 2021, 12, 918. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Qiu, Z.; Yuan, H.; Wang, X.; Li, X.; Sun, H.; Guo, X.; Lu, Y.; Feng, X.; Majid, M.; et al. Evolutionary rates of and selective constraints on the mitochondrial genomes of Orthoptera insects with different wing types. Mol. Phylogen. Evol. 2020, 145, 106734. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, J.; Xue, X.; Qin, G.; Gao, Y.; Li, K.; Zhang, Y.; Li, X.J. Comparative analysis of mitogenomes among three species of grasshoppers (Orthoptera: Acridoidea: Gomphocerinae) and their phylogenetic implications. PeerJ 2023, 11, e16550. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.L.; Farr, C.L.; Farquhar, A.L.; Kaguni, L.S. Sequence, organization, and evolution of the A+T region of Drosophila melanogaster mitochondrial DNA. Mol. Biol. Evol. 1994, 11, 523–538. [Google Scholar] [CrossRef]
- Brehm, A.; Harris, D.J.; Hernández, M.; Cabrera, V.M.; Larruga, J.M.; Pinto, F.M.; González, A.M. Structure and evolution of the mitochondrial DNA complete control region in the Drosophila subobscura subgroup. Insect Mol. Biol. 2001, 10, 573–578. [Google Scholar] [CrossRef]
- Ma, Y.; Miao, Y. Mitogenomic comparison of the mole crickets Gryllotalpidae with the phylogenetic implications (Orthoptera: Ensifera). Insects 2022, 13, 919. [Google Scholar] [CrossRef]
- Pereira, R.J.; Ruiz-Ruano, F.J.; Thomas, C.J.; Pérez-Ruiz, M.; Jiménez-Bartolomé, M.; Liu, S.; de la Torre, J.; Bella, J.L. Mind the numt: Finding informative mitochondrial markers in a giant grasshopper genome. J. Zool. Syst. Evol. Res. 2021, 59, 635–645. [Google Scholar] [CrossRef]
- Tsuji, J.; Frith, M.C.; Tomii, K.; Horton, P. Mammalian NUMT insertion is non-random. Nucleic Acids Res. 2012, 40, 9073–9088. [Google Scholar] [CrossRef] [PubMed]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenkoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Proutski, V.; Holmes, E. SWAN: Sliding window analysis of nucleotide sequence variability. Bioinformatics 1998, 14, 467–468. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 7–3022. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Schwarz, G. The bayesian information criterion. Ann. Stat. 1978, 6, 461–464. [Google Scholar]
- Bey-Bienko, G.Y.; Mistshenko, L.L. Locusts and Grasshoppers of the USSR and Adjacent Countries; Part II; Academy of Sciences of USSR Publisher: Moscow-Leningrad, Russia, 1951; pp. 1–287. (In Russian) [Google Scholar]
- Latchininsky, A.V.; Sergeev, M.G.; Childebaev, M.K.; Chernjakhovsky, M.E.; Lockwood, J.A.; Kambulin, V.E.; Gapparov, F.A. Grasshoppers of Kazakhstan, Middle Asia and Adjacent Territories; Association for Applied Acridology International, University of Wyoming: Laramie, WY, USA, 2002; 387p. (In Russian) [Google Scholar]
- Berger, D. The Evolution of Complex Courtship Songs in the Genus Stenobothrus Fischer, 1853 (Orthoptera, Caelifera, Gomphocerinae). Ph.D. Thesis, Friedrich-Alexander-Universität Erlangen, Erlangen, Germany, 2008; 169p. [Google Scholar]
- Berger, D.; Chobanov, D.P.; Mayer, F. Interglacial refugia and range shifts of the alpine grasshopper Stenobothrus cotticus (Orthoptera: Acrididae: Gomphocerinae). Org. Divers. Evol. 2010, 10, 123–133. [Google Scholar] [CrossRef]
- Gantigmaa, C.; Myagmar, G. Orthopteroid Insects in Mongolia; Mongolian Academy of Sciences: Ulaanbaatar, Mongolia, 2022; 564p. (In Mongolian) [Google Scholar]
- Kenyeres, Z.; Bauer, N.; Kociński, M.; Grzywacz, B. Genetic and morphological differences among relict marginal occurrences of Stenobothrus eurasius (Orthoptera). Arthropod Syst. Phylogen. 2024, 82, 503–514. [Google Scholar] [CrossRef]
- Sergeev, M.G. Distribution Patterns of Grasshoppers and Their Kin over the Eurasian Steppes. Insects 2021, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zheng, Z.M.; Huang, Y.; Zhou, Z.J.; Wang, L. Comparative analysis of the mitochondrial control region in Orthoptera. Zool. Stud. 2011, 50, 385–393. [Google Scholar]
- Papadopoulou, A.; Anastasiou, I.; Vogler, A.P. Revisiting the insect mitochondrial molecular clock: The mid-Aegean trench calibration. Mol. Biol. Evol. 2010, 27, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Zhongying, Q.; Huihui, C.; Hao, Y.; Yuan, H.; Huimeng, L.; Xia, L.; Xingchun, G. Comparative mitochondrial genomes of four species of Sinopodisma and phylogenetic implications (Orthoptera, Melanoplinae). ZooKeys 2020, 969, 23. [Google Scholar] [CrossRef]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication origin of mitochondrial DNA in insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Intern. Rev. Cyt. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Zinovkina, L.A. DNA replication in human mitochondria. Biochemistry 2019, 84, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Brower, A.V. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Nat. Acad. Sci. USA 1994, 91, 6491–6495. [Google Scholar] [CrossRef]
- Pons, J.; Vogler, A.P. Complex pattern of coalescence and fast evolution of a mitochondrial rRNA pseudogene in a recent radiation of tiger beetles. Mol. Biol. Evol. 2005, 22, 991–1000. [Google Scholar] [CrossRef]
- Avise, J.C.; Walker, D.E. Pleistocene phylogeographic effects on avian populations and the speciation process. Proc. R. Soc. Lond. Ser. B 1998, 265, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Nattier, R.; Robillard, T.; Amedegnato, C.; Couloux, A.; Cruaud, C.; Desutter–Grandcolas, L. Evolution of acoustic communication in the Gomphocerinae (Orthoptera: Caelifera: Acrididae). Zool. Scr. 2011, 40, 479–497. [Google Scholar] [CrossRef]
- Berger, D.; Gottsberger, B. Analysis of the courtship of Myrmeleotettix antennatus (Fieber, 1853) –With general remarks on multimodal courtship behavior in gomphocerine grasshoppers. Articulata 2010, 25, 1–21. [Google Scholar]
- Vedenina, V.; Sevastianov, N.; Kovalyova, E. New data on bioacoustics and courtship behaviour in grasshoppers (Orthoptera, Acrididae, Gomphocerinae) from Russia and adjacent countries. ZooKeys 2024, 1200, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Nolen, Z.J.; Yildirim, B.; Irisarri, I.; Liu, S.; Groot Crego, C.; Amby, D.B.; Mayer, F.; Gilbert, M.T.P.; Pereira, R.J. Historical isolation facilitates species radiation by sexual selection: Insights from Chorthippus grasshoppers. Mol. Ecol. 2020, 29, 4985–5002. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Dufresnes, C.; Krištín, A.; Künzel, S.; Vences, M.; Hawlitschek, O. Phylogenetic insights into Central European Chorthippus and Pseudochorthippus (Orthoptera: Acrididae) species using ddRADseq data. Mol. Phylogen. Evol. 2024, 193, 108012. [Google Scholar] [CrossRef] [PubMed]
- Berdan, E.L.; Mazzoni, C.J.; Waurick, I.; Roehr, J.T.; Mayer, F. A population genomic scan in Chorthippus grasshoppers unveils previously unknown phenotypic divergence. Mol. Ecol. 2015, 24, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J.F.D.; Mendes, C.C.; Blogg, A.; Payne, A.; Tanaka, K.M.; Gaspar, P.; Jimenez, J.F.; Kittelmann, M.; McGregor, A.P.; Nunes, M.D.S. Tartan underlies the evolution of Drosophila male genital morphology. Proc. Natl. Acad. Sci. USA 2019, 116, 19025–19030. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, A.M.; Hood, E.J.; Jimenez, J.F.; Nunes, M.D.S.; McGregor, A.P. Sox21b underlies the rapid diversification of a novel male genital structure between Drosophila species. Current Biology 2024, 34, 1114–1121. [Google Scholar] [CrossRef]
- Gupta, K.; Dhawan, R.; Kajla, M.; Misra, T.; Kumar, S.; Gupta, L. The evolutionary divergence of STAT transcription factor in different Anopheles species. Gene 2017, 596, 89–97. [Google Scholar] [CrossRef]
- Horsák, M.; Chytrý, M.; Hájková, P.; Hájek, M.; Danihelka, J.; Horsáková, V.; Horsáková, V.; Ermakov, N.; German, D.A.; Kočí, M.; et al. European glacial relict snails and plants: Environmental context of their modern refugial occurrence in southern Siberia. Boreas 2015, 44, 638–657. [Google Scholar] [CrossRef]
- Kühne, G.; Kosuch, J.; Hochkirch, A.; Schmitt, T. Extra-Mediterranean glacial refugia in a Mediterranean faunal element: The phylogeography of the chalk-hill blue Polyommatus coridon (Lepidoptera, Lycaenidae). Sci. Rep. 2017, 7, 43533. [Google Scholar] [CrossRef]
- Pavelková Řičánková, V.; Robovský, J.; Riegert, J. Ecological structure of recent and last glacial mammalian faunas in northern Eurasia: The case of Altai-Sayan refugium. PLoS ONE. 2014, 9, e85056. [Google Scholar] [CrossRef]
- Stewart, J.R.; Lister, A.M.; Barnes, I.; Dalén, L. Refugia revisited: Individualistic responses of species in space and time. Proc. R. Soc. B Biol. Sci. 2010, 277, 661–671. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Stöck, M.; Zhang, P.; Wang, X.L.; Zhou, H.; Qu, L.H. Phylogeography of a widespread terrestrial vertebrate in a barely-studied Palearctic region: Green toads (Bufo viridis subgroup) indicate glacial refugia in Eastern Central Asia. Genetica 2008, 134, 353–365. [Google Scholar] [CrossRef]
- Barth, A.M.; Clark, P.U.; Bill, N.S.; He, F.; Pisias, N.G. Climate evolution across the Mid-Brunhes transition. Clim. Past. 2018, 14, 2071–2087. [Google Scholar] [CrossRef]
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Zhang, X.; Cui, Y.; Liu, L.; Wang, X.; Gou, X. Characterization of the complete mitochondrial genome of Euchorthippus unicolor (Orthoptera: Arcypteridae). Mitochondrial DNA Part. B 2019, 4, 846–847. [Google Scholar] [CrossRef]
- Chapco, W.; Contreras, D. Subfamilies Acridinae, Gomphocerinae and Oedipodinae are “fuzzy sets”: A proposal for a common African origin. J. Orthoptera Res. 2011, 20, 173–190. [Google Scholar] [CrossRef]
- Laiolo, P.; Illera, J.C.; Obeso, J.R. Local climate determines intra-and interspecific variation in sexual size dimorphism in mountain grasshopper communities. J. Evol. Biol. 2013, 26, 2171–2183. [Google Scholar] [CrossRef] [PubMed]
- Sirin, D.; Taylan, M.S.; Bircan, R.; Akyildiz, G.; Can, L. Phylogenetic and phylogeographic analysis of Myrmeleotettix maculatus (Orthoptera: Acrididae: Gomphocerinae) species group in Anatolia. Zootaxa 2021, 4949, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, A.; Mao, S.; Huang, Y. DNA barcoding and species boundary delimitation of selected species of Chinese Acridoidea (Orthoptera: Caelifera). PLoS ONE 2013, 8, e82400. [Google Scholar] [CrossRef] [PubMed]
- Contreras, D.; Chapco, W. Molecular phylogenetic evidence for multiple dispersal events in gomphocerine grasshoppers. J. Orthoptera Res. 2006, 15, 91–98. [Google Scholar] [CrossRef]
- Laiolo, P.; Pato, J.; Illera, J.C.; Obeso, J.R. Selection for functional performance in the evolution of cuticle hardening mechanisms in insects. Evolution 2021, 75, 1132–1142. [Google Scholar] [CrossRef]
- Bugrov, A.; Novikova, O.; Mayorov, V.; Adkison, L.; Blinov, A. Molecular phylogeny of Palaearctic genera of Gomphocerinae grasshoppers (Orthoptera, Acrididae). Syst. Entomol. 2006, 31, 362–368. [Google Scholar] [CrossRef]
- Zhao, L.; Zhan, L.; Li, Q.; Ren, J. DNA barcoding of nymph grasshoppers from Xinjiang. Xinjiang Agric. Sci. 2015, 52, 785. (In Chinese) [Google Scholar]
- Song, H.; Béthoux, O.; Shin, S.; Donath, A.; Letsch, H.; Liu, S.; McKenna, D.D.; Meng, G.; Misof, B.; Podsiadlowski, L.; et al. Phylogenomic analysis sheds light on the evolutionary pathways towards acoustic communication in Orthoptera. Nat. Commun. 2020, 11, 4939. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Region | COI Gene | k CR/COI | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | S | PIS | p-Distance ± SE | T3P (CR + tRNA) ± SE | T3P (CR) | H | S | PIS | p-Distance ± SE | T3P ± SE | ||
| Stenobothrus/M. palpalis | 30/2 | 52 | 42 | 4.89 ± 0.79 | 5.13 ± 0.90 | 6.58 ± 1.08 | 41/4 | 60 | 43 | 5.18 ± 0.89 | 5.41 ± 1.05 | 1.22 |
| Stenobothrus/M. maculatus group * | 30/3 | 64 | 43 | 5.34 ± 0.78 | 5.63 ± 0.87 | 7.00 ± 1.14 | 41/9 | 83 | 53 | 6.59 ± 0.88 | 7.03 ± 1.07 | 1.00 |
| Stenobothrus/ Omocestus | 30/3 | 58 | 45 | 5.03 ± 0.72 | 5.26 ± 0.84 | 6.61 ± 1.12 | 41/11 | 82 | 57 | 6.91 ± 0.94 | 7.26± 1.17 | 0.91 |
| Stenobothrus/C. pullus | 30/1 | 72 | – | 8.24 ± 1.15 | 9.00 ± 1.38 | 11.50 ± 1.62 | 41/1 | 64 | – | 6.35 ± 1.06 | 6.75 ± 1.11 | 1.7 |
| Stenobothrus/P. parallelus | 30/1 | 88 | – | 11.08 ± 1.20 | 12.43 ± 1.57 | 16.50 ± 2.11 | 41/1 | 70 | – | 8.44 ± 1.14 | 9.13 ± 1.36 | 1.8 |
| Stenobothrus/G. rufus | 30/1 | 94 | – | 12.56 ± 1.27 | 14.14 ± 1.70 | 17.79 ± 2.19 | 41/1 | 75 | – | 8.94 ± 1.12 | 9.68 ± 1.35 | 1.83 |
| Stenobothrus/ Euchorthippus | 30/1 | 119 | – | 15.88 ± 1.47 | 18.48 ± 1.86 | 22.84 ± 2.61 | 41/1 | 95 | – | 13.23 ± 1.33 | 14.75 ± 1.65 | 1.54 |
| Control Region | COI Gene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H | S | PIS | p-Distance ± SE | T3P (CR + tRNA) ± SE | T3P (CR) | H | S | PIS | p-Distance ± SE | T3P ± SE | |
| S. eurasius | 12 | 10 | 8 | 0.59 ± 0.21 | 0.60 ± 0.21 | 0.79 ± 0.28 | 17 | 35 | 14 | 1.47 ± 0.33 | 1.49 ± 0.28 |
| S. hyalosuperficies | 8 | 14 | 4 | 0.74 ± 0.22 | 0.74 ± 0.22 | 0.99 ± 0.27 | 3 | 11 | 0 | 1.34 ± 0.42 | 1.36 ± 0.43 |
| Stenobothrus | 30 | 24 | 17 | 0.87 ± 0.23 | 0.88 ± 0.22 | 1.03 ± 0.3 | 41 | 41 | 22 | 1.27 ± 0.25 | 1.29 ± 0.28 |
| S. eurasius Altai | S. eurasius Contact Zone | S. hyalosuperficies Kazakhstan | S. hyalosuperficies Contact Zone | S. hyalosuperficies | |
|---|---|---|---|---|---|
| S. eurasius Altai | 0.17 ± 0.08 | 0.45 ± 0.13 | 0.46 ± 0.16 | 0.43 ± 0.16 | 0.52 ± 0.17 |
| S. eurasius contact zone | 45.7 ± 15.6 | 0.32 ± 0.12 | 0.39 ± 0.16 | 0.37 ± 0.11 | 0.41 ± 0.12 |
| S. hyalosuperficies Kazakhstan | 45.57 ± 15.63 | 18.24 ± 9.61 | 0.32 ± 0.14 | 0.428 ± 0.123 | – |
| S. hyalosuperficies contact zone | 32.56 ± 12.96 | 1.34 ± 3.58 | 13.82 ± 8.77 | 0.41 ± 0.13 | – |
| S. hyalosuperficies | 42.43 ± 11.69 | 8.57 ± 3.58 | – | – | 0.43 ± 0.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorokina, S.; Sevastianov, N.; Tarasova, T.; Vedenina, V. The Fast Evolution of the Stenobothrini Grasshoppers (Orthoptera, Acrididae, and Gomphocerinae) Revealed by an Analysis of the Control Region of mtDNA, with an Emphasis on the Stenobothrus eurasius Group. Insects 2024, 15, 592. https://doi.org/10.3390/insects15080592
Sorokina S, Sevastianov N, Tarasova T, Vedenina V. The Fast Evolution of the Stenobothrini Grasshoppers (Orthoptera, Acrididae, and Gomphocerinae) Revealed by an Analysis of the Control Region of mtDNA, with an Emphasis on the Stenobothrus eurasius Group. Insects. 2024; 15(8):592. https://doi.org/10.3390/insects15080592
Chicago/Turabian StyleSorokina, Svetlana, Nikita Sevastianov, Tatiana Tarasova, and Varvara Vedenina. 2024. "The Fast Evolution of the Stenobothrini Grasshoppers (Orthoptera, Acrididae, and Gomphocerinae) Revealed by an Analysis of the Control Region of mtDNA, with an Emphasis on the Stenobothrus eurasius Group" Insects 15, no. 8: 592. https://doi.org/10.3390/insects15080592
APA StyleSorokina, S., Sevastianov, N., Tarasova, T., & Vedenina, V. (2024). The Fast Evolution of the Stenobothrini Grasshoppers (Orthoptera, Acrididae, and Gomphocerinae) Revealed by an Analysis of the Control Region of mtDNA, with an Emphasis on the Stenobothrus eurasius Group. Insects, 15(8), 592. https://doi.org/10.3390/insects15080592