1. Introduction
The baculovirus expression system (BES), which utilizes entomopathogenic baculoviruses and insect cells, is a highly valuable system for the production of various recombinant proteins [
1]. Compared to other host-based expression systems, the BES offers several advantages, including cost-effectiveness, biosafety for researchers, and the ability to provide post-translational modifications of recombinant proteins similar to those found in mammalian expression systems [
2]. For the efficient production of target proteins in a BES, it is crucial to establish optimal viral infection conditions [
3]; therefore, optimizing the multiplicity of infection (MOI)—the degree of viral infection—is essential to ensure the efficient production of target proteins by maintaining a balance between cell viability and virus production [
4].
Accurate virus titration is essential for optimizing the MOI during virus infection. Various virus titration methods, including plaque assays [
5], end-point dilution assays involving TCID
50 (tissue culture infective dose 50) [
6], quantitative PCR (qPCR) [
7], flow cytometry [
8], and enzyme-linked immunosorbent assays (ELISAs) [
9], have been developed and are widely used; however, each method has its limitations, such as specific equipment requirements, technical difficulties, or accuracy issues related to experimenter proficiency. Among these methods, the end-point dilution assay involving TCID
50 is the most commonly used. This method calculates the virus titer by performing serial dilutions of the virus and infecting insect cells, followed by determining the ratio of infected to uninfected wells [
10]. This method also allows for accurate virus titer determination even at very low virus concentrations and provides statistically reliable data; however, because this method statistically determines virus titers based on the observation of the cytopathic effect (CPE), it relies on visual observation to determine infection, which introduces subjective interpretation by experimenters, leading to potential variability in the results [
6].
To overcome the limitations of the described methods, a transgenic cell line, Sf9-ET, was developed based on the Sf9 insect cell line, which can express enhanced green fluorescent protein (EGFP) upon infection with a baculovirus [
11]. This allowed for the relatively easy identification of virus infections under fluorescence microscopy compared to the CPE. Nevertheless, challenges remained, including the long time required for titration due to low fluorescence expression. Hence, the Sf9-QE transgenic cell line was developed to overcome this shortcoming [
12]. Sf9-QE cells utilize a combination of regulatory elements, including the homologous region 3 (hr3),
39k promoter, and
p10 promoter, for a more efficient expression of EGFP. As a result, Sf9-QE cells can complete titration earlier than Sf9-ET cells due to the rapid and strong expression of EGFP; however, the final virus titer determination still requires 6.0 to 6.3 days.
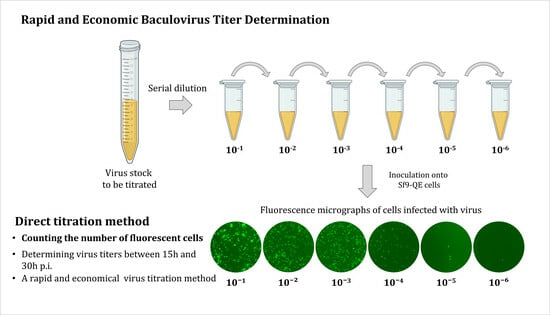
Therefore, in this study, we aimed to develop a new titration method, the direct titration method, with a dramatically reduced time required to determine titers using the Sf9-QE cell line (
Figure 1). In the plaque assay, the virus is directly titrated by the number of plaques formed after serial dilution and the inoculation of the virus to be titrated [
5]. Using the same principle, our hypothesis was that virus titer determination would be possible if the number of virus-infected cells could be directly counted after serial dilutions of the virus to be titrated; however, unlike the plaque assay, reliable virus titer determination was expected to be possible only if the phenotype identified with EGFP during the primary infection in Sf9-QE cells could be confirmed before the phenotype after the secondary infection of the virus. The observation of EGFP expression in the Sf9-QE cells as early as one day post-infection (d.p.i.) showed the feasibility of such virus titer determination [
12]. It has been reported that the budding of baculoviruses occurs from about 17 to 20 h after primary virus infection, with the infection of other cells and the expression of the phenotype occurring after additional time [
13]. The rapid and robust expression of the phenotype is required to confirm the phenotype of the primary infection of the virus prior to the expression of the phenotype of the cells after the secondary infection. In the case of Sf9-QE, the early expression of EGFP has already been demonstrated as early as one day after viral infection by the
39k promoter, a delayed-early promoter that is activated by the immediately early 1 (IE–1) protein and operates from 3 to 6 h after infection [
14,
15]. Additionally, although the rapid expression of EGFP at 1 d.p.i. was confirmed, it was believed that the copy number of the transgene present in the transfected cell line would have a major influence on the quantitative expression; therefore, the determination of the copy number of the EGFP transgene in the Sf9-QE cells would further confirm the feasibility of developing a rapid titration method using Sf9-QE cells.
This study presents a new direct titration method that utilizes the distinct properties of the Sf9-QE cell line to overcome the limitations of traditional virus titration techniques through a cost-effective, rapid process while minimizing subjectivity.
2. Materials and Methods
2.1. Cells and Viruses
The
Spodoptera frugiperda cell line Sf9 and the transgenic cell line Sf9-QE were cultured at 27 °C using an Sf–900™ II SFM medium (Gibco, Grand Island, NY, USA) and an Sf–900™ II SFM medium supplemented with G418 (Invitrogen, Carlsbad, CA, USA), respectively. The viruses used consisted of rMultibac, rAc-polh-TEVp, rAc-polh-CVA6-P1, and rAc-polh-RSV-F. rMultibac was generated using Multibac (Geneva Biotech, Geneva, Switzerland), a bacmid derived from
Autographa californica nucleopolyhedrovirus (AcMNPV-E2, GenBank No. KM667940.1). rAc-polh-TEVP, rAc-polh-CVA6-P1, and rAc-polh-RSV-F are recombinant viruses that express the protease of the tobacco etch virus, the P1 protein of Coxsackievirus A6, and the fusion (F) protein of the respiratory syncytial virus under a
polyhedrin promoter, respectively (
Figure S1).
2.2. Cell Genomic DNA Extraction
Genomic DNA extraction from Sf9-QE cells was performed using an AccuPrep Genomic DNA Extraction Kit (Bioneer, Daejeon, Republic of Korea) according to the manufacturer’s instructions. Briefly, cells were collected by centrifugation at 4000 rpm for 5 min, and the supernatant was discarded. The cell pellet was washed with PBS (1 X) and centrifuged under the same conditions. After removing the supernatant, the cell pellet was resuspended in PBS (1 X). Cells were lysed by adding the provided lysis buffer, followed by the addition of absolute ethanol to the lysate. The mixture was transferred to a spin column and centrifuged at 8000 rpm for 1 min. The column membrane was washed with the provided wash buffer and subjected to a final centrifugation at 13,000 rpm for 1 min to remove the remaining ethanol. The column was then placed into a new microcentrifuge tube, and 50 µL of nuclease-free water was added. The column was centrifuged at 8000 rpm for 1 min to elute the genomic DNA.
2.3. Quantitative Real-Time PCR
To estimate the copy number of the transgene, qPCR was conducted using a QuanStudio1 Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) with Power SYBR™ Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Genomic DNA extracted from Sf9-QE cells was serially diluted 2-fold to generate five different concentrations. Each qPCR reaction was performed in a total volume of 20 µL, consisting of 1 µL of template DNA from each concentration, 10 µL of Power SYBR™ Green PCR Master Mix, 1 µL each of forward and reverse primer for the amplification of the EGFP transgene and reference genes, and nuclease-free water to adjust the final volume. Amplification was performed with an initial denaturation at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 1 s and annealing at 60 °C for 30 s, and a melt-curve analysis was conducted after amplification at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s. Each reaction was performed in triplicate.
2.4. End-Point Dilution Assay Involving TCID50
To perform the end-point dilution assay involving TCID
50 based on the Reed–Muench method [
6], Sf9-QE cells were distributed at a density of 2.0 × 10
4 cells/90 µL per well in a 96-well cell culture plate. The virus was serially diluted using an Sf–900™ II SFM medium, and 10 µL of the virus at each dilution was inoculated into 8 wells of distributed cells. Then, cells were incubated under the same conditions as described above. Six days after virus inoculation, the number of infected and uninfected wells was counted for each virus dilution using a fluorescence microscope, and the virus titer was calculated using TCID
50.
2.5. Direct Titration Method
Sf9-QE cells were distributed at a density of 2.0 × 104 cells/90 µL per well in a 96-well cell culture plate. The virus was serially diluted using an Sf–900™ II SFM medium, and 10 µL of the virus at each dilution was inoculated into the 3 wells (for 3 repetitions) of distributed cells. Then, the cells were incubated under the same conditions as described above, and the number of cells expressing EGFP was counted using a fluorescence microscope at specific times after infection. The virus titer was calculated by multiplying the average number of cells counted and the viral dilution ratio.
2.6. Statistical Analysis
All statistical analyses were performed using IBM SPSS Statistics version 25 (IBM Corp., Armonk, NY, USA). Descriptive statistics were calculated for each group, including means and standard deviations. To evaluate the significance of differences between groups, a t-test was conducted on triplicate experimental groups. A p-value of less than 0.05 was considered statistically significant.
4. Discussion
In this study, a novel direct titration method for virus titer determination was developed that is convenient and enables accurate titration in a short period of time—within 24 h—using unique Sf9-QE cells. Similarly to other existing virus titration methods, the direct titration method showed that virus titers can be determined by serially diluting the virus, inoculating each dilution onto 1 well of a 96-well plate, and counting infected cells before and after 24 h p.i. The reason for this convenient and rapid titration was the use of Sf9-QE cells; transgenic Sf9-QE cells can only express fluorescence upon viral infection, making it convenient to check for viral infection. In addition, the expression of the EGFP gene for fluorescence is achieved by utilizing both the
39k promoter and strong
p10 promoter, which start to activate about 3 to 6 h p.i., allowing for rapid and high expression. Furthermore, we confirmed the presence of at least seven copies of the EGFP transgene in Sf9-QE cells, indicating that higher levels of EGFP expression may be possible. The production of a specific recombinant protein using a BES typically takes at least four–eight weeks from the generation of the recombinant virus to the identification of the target protein. Various improvements have been made to methods such as recombinant virus generation, isolation, and titration to shorten the time required to produce recombinant proteins [
18,
19,
20]. In particular, virus titration takes at least 1 to 10 days to complete, depending on the method. Typical titration methods that can be performed in as little as one to two days include qPCR, ELISAs, and flow cytometry [
7,
8,
9,
21,
22]; however, these methods have disadvantages, such as the accuracy of titration depending on the experimenter, the complexity of the experimental process, and high cost. Therefore, if it is possible to determine virus titers in a short time with a simple experimental process and low cost, it will make the BES easier and cheaper to use.
The hypothesis for this study was confirmed by the fact that accurate virus titration was possible within 15 to 30 h p.i., and that titration after 33 h p.i. was not accurate. Theoretically, given the formation time of BVs as a secondary infectious agent (17–20 h p.i.) and the early expression time of EGFP (3–6 h p.i.), fluorescence expression by secondary infection could be possible at 20–23 h p.i.; however, in our results, the accurate quantification of virus titers by direct titration was possible up to 30 h p.i. These results are believed to indicate that more time is required for the secondary infection of the virus and for verifiable levels of fluorescence to be visibly expressed. Although the accuracy of the direct titration method for determining virus titers was demonstrated in this study using repeated experiments and a variety of virus samples, it is not clear whether these results are the result of counting only fluorescent cells from primary infection, as we hypothesized.
For the direct counting of fluorescent Sf9-QE cells via primary infection with the virus, the culture status of the cells was determined to be a very important factor. Therefore, we compared the accuracy of virus titer determination by the direct titration method according to the culture period after subculturing, and showed that cells cultured within 3 days should be used. These results not only suggest that the proliferation of viruses is affected by the proliferation status of cells, but also show that a proper cell condition is required for virus titer determination by the direct titration method. According to a previous report, the proliferation of Sf9-QE cells remains in the log phase for about five to six days and then reaches the stationary phase [
12]. Therefore, even Sf9-QE cells cultured for three and five days in the log phase have different effects on the proliferation of viruses, resulting in different virus titers determined by the direct titration method. Furthermore, for the direct counting of fluorescent Sf9-QE cells, counting too many cells is not only impractical, but also time-consuming. Therefore, we explored appropriate observation dilution ratios depending on the concentration of virus to be titrated and found that the concentration of virus to be titrated does not affect virus titer determination by the direct titration method. We also found that virus quantification values are reliable even at high virus dilution ratios with a minimum count of 10 or less. These results indicate that virus titer determination by the direct titration method is easier and faster; however, in our experimental experience, the selection of cells to be counted by direct titration requires some judgment on the part of the experimenter as to whether it is more appropriate to count cells at a viral dilution of 10 or less or at the dilution immediately preceding that dilution. In some cases, counting at both dilutions may be a good option.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






