Abstract
To theoretically examine the structural transition of vacancy–solute complexes in Al–Mg–Si alloys, we performed first-principles calculations for layered vacancy–solute complexes with additional Mg atoms. The central Mg atom in the additional Mg layer shifted to the Si layer with the increase in the number of Mg atoms to weaken the repulsive Mg–Mg interaction and to form Mg–Si bonds. When five Mg atoms were added to the layered vacancy–solute complex, the central Mg atom completely shifted to the Si layer, and a Mg vacancy was formed in the Mg layer, which indicated that the β″-eye is formed upon the addition of Mg atoms. We reproduced β″-eye formation from a solid solution with a vacancy using first-principles-based Monte Carlo simulations. Once the β″-eye was formed on the layered vacancy–solute complex, the process can be repeated by the formation of alternate Mg and Si layers along (010) β″. These results clearly indicate that the layered vacancy–solute complex plays an important role in β″-eye formation.
1. Introduction
The use of 6000 series Al–Mg–Si alloys for manufacturing automobile body panels has increased because such alloys exhibit hardening characteristics. The precipitation of the β″ phase during paint-bake treatment is mainly responsible for the hardening in Al–Mg–Si alloys [1,2,3]. However, the solute clusters formed during storage at room temperature, referred to as natural aging, suppresses the precipitation of the β″ phase [4,5,6]. In our recent study [7], we determined the stable structures of vacancy–solute complexes, V(Mg,Si)n for n = 1–12, in Al–Mg–Si alloys using first-principles calculations and predicted the initial structures through multiple linear regression analysis. We observed that the formation energy of the vacancy–solute complex decreased with the increasing number of solute atoms, and the VMg4Si8 complex exhibited the lowest formation energy.
Regarding the β″ phase, theoretical studies have been carried out to investigate its structural properties [8,9,10,11,12,13,14], chemical bonding [8,9,14], and stability [10,11,13,14]. However, the structural transition to the β″ phase remains unclear. The formation of a layered MgSi phase in an Al matrix was reported using a multiscale approach [15]. However, the formation of the β″ phase, which requires a shift of Mg atoms to the Si layer, was not observed. Before we proceed to investigate the structural transition from the vacancy–solute complexes to the β″ phase, it is important to clarify the relationship between the vacancy–solute complexes and the β″-eye, which is the precursor of the β″-phase [2]. In this study, to investigate the structural transition from the vacancy–solute complexes to the β″-eye in Al–Mg–Si alloys, we performed first-principles calculations for the vacancy–solute complexes with additional Mg atoms in Al–Mg–Si alloys. To reproduce the structural transition of the vacancy–solute complexes from the solid solution in the Al–Mg–Si alloy, we performed first-principles-based Monte Carlo (MC) simulations.
2. Computational Methods
The atomic arrangement of the (001) plane of the Al matrix and the (010) plane of β″-Mg5Si6 [1,16] is shown in Figure 1a. The frame of the unit cell of β″-Mg5Si6 was fitted to the Al matrix. The number of atoms in the unit cell of β″-Mg5Si6 was the same as that in the adjacent area in the Al matrix. However, the Mg atom at the corner of the unit cell of β″-Mg5Si6 was located not on an face-centered cubic (FCC) lattice site but on an octahedral site of FCC as shown in the side view around the β″-eye in β″-Mg5Si6 in Figure 1b. Instead, an FCC lattice site in the middle of the a-axis of β″-Mg5Si6 is vacant. The octahedral site occupied by the Mg atom corresponds to the vacant site in the adjacent (010) plane. Therefore, considering its atomic arrangement, β″-Mg5Si6 can be considered as a structure in which an FCC lattice site occupied by a Mg atom is shifted to an octahedral site surrounded by four Si atoms in the adjacent plane. As a result of the shift of the Mg atom, the four Si atoms around the Mg atom are outwardly displaced, and the four Mg atoms around the vacant site are displaced inward from the ideal lattice sites of the Al matrix. The driving forces of the Mg atom shift possibly are the energy gain by the formation of Mg–Si bonds and the reduction of repulsive Mg–Mg interaction, as will be discussed later.
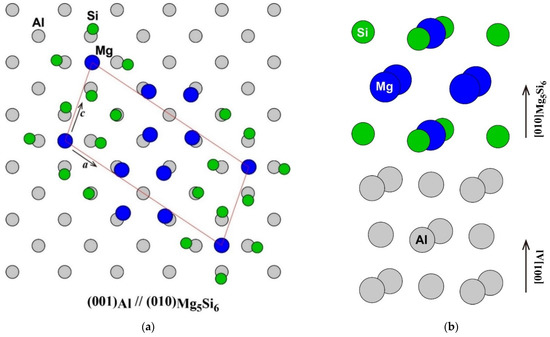
Figure 1.
(a) Atomic arrangement of the (001) plane of Al matrix and the (010) plane of β″-Mg5Si6 (a) and (b) the side view around the β″-eye in β″-Mg5Si6.
Figure 2 shows the structures of the most stable and second most stable VMg4Si8 complexes obtained in our previous study [7] and the β″-eye in β″-Mg5Si6; the second most stable VMg4Si8 complex exhibits a layered structure. The structural feature of the β″-eye is the presence of Mg atoms in the Si layer. The comparison of the structures of the layered VMg4Si8 complex, and the β″-eye indicates that the length of the Si–Si bond in the β″-eye is ≈1 Å longer than that in the layered VMg4Si8 complex, while the lengths of the Mg–Si and Mg–Mg bonds are identical. This is because the Si-Si bonds must be expanded to accommodate the Mg–Si bonds in the Si layer. To examine the structural transition of the layered VMg4Si8 complex due to the addition of Mg atoms, we performed the first-principles calculations for the layered VMg4Si8 + Mgn complexes.

Figure 2.
Structures of (a) the most stable, (b) second most stable VMg4Si8 complexes, and (c) the β″-eye in β″-Mg5Si6.
We employed the same size of the supercell that was used to calculate the formation energies of VMg4Si8 complexes [7]: a supercell composed of 108 lattice sites with an FCC structure (3 × 3 × 3 unit cell). The length of the supercell was determined using the calculated equilibrium lattice parameter of the aluminum matrix, 4.0396 Å, which is in good agreement with the experimental value of 4.0496 Å. We employed the Vienna ab initio simulation package (VASP) [17,18] along with the generalized gradient approximation of Perdew–Burke–Ernzerhof (PBE) [19]. Potentials based on the all-electron projector augmented wave (PAW) method were also used [20,21]. All calculations were performed with a 350 eV energy cutoff and a 4 × 4 × 4 k mesh in the Monkhorst–Pack scheme. The structural relaxation of the atoms was continued until the total energy change decreased below 0.001 eV. The supercell volume and shape parameters were fixed for consistency with the clustering behavior of the solute atoms: during solute cluster formation, both the solute atom concentration and matrix lattice parameter remained essentially unchanged.
The formation energy of the VMgmSin complex, , can be obtained as follows (with reference to an isolated vacancy and isolated solute atoms):
where is the total energy of the supercell with a VMgmSin complex, is the total energy of the supercell of the perfect lattice, , and are the total energies of the supercell with a vacancy, a Mg atom, and a Si atom, respectively.
The binding energy of the Mg atom added to the VMg4Si8 + Mgn−1 complex is defined as follows:
Negative (positive) values indicate energy gain (penalty).
To examine the reproduction of the vacancy–solute complexes from the solid solution in the Al–Mg–Si alloy, we performed the first-principles-based MC simulations. The temperature in the MC simulations was set at 200 K to avoid an excessive increase in the potential energy (shown in Appendix A, Figure A1). Metropolis–Hastings sampling was employed to determine the acceptance probability of the position swap [22]. The energy during the MC simulations was calculated using a k mesh reduced to a 2 × 2 × 2 k mesh.
3. Results and Discussion
Figure 3 shows the formation energies of VMg4Si8 + Mgn (n = 1–5) complexes and the binding energy of a Mg atom to the VMg4Si8 + Mgn−1 complex. For comparison, the calculated results for the most stable VMg4Si8 + Mg complex are shown in Figure 3, and the structures of the VMg4Si8 + Mgn (n = 1–5) complexes are shown in Figure 4. The formation energy of VMg4Si8 + Mg1 decreased as a result of the formation of four Mg–Si bonds. In Al–Mg–Si alloys, the Mg–Si bond possesses the lowest interaction energy around a vacancy and in the Al matrix [7]. In the VMg4Si8 + Mg2 complex, both Mg–Si and Mg–Mg bonds were formed upon the addition of Mg atoms. The Mg–Mg interaction is repulsive in the Al matrix [7]. Thus, the formation energy of the VMg4Si8 + Mgn complexes for n = 2–4 slightly increased owing to the positive interaction energy of the Mg–Mg bonds. However, the total formation energy of these complexes still decreased because the binding energy of the Mg atom to the VMg4Si8 + Mgn−1 complex is negative. There is a steep drop both in the formation energy and the binding energy of the VMg4Si8 + Mg5 complex. All possible sites in the most stable VMg4Si8 complex with additional Mg were examined; the calculated results of the most stable VMg4Si8 + Mg are shown in Figure 3. In the most stable VMg4Si8 + Mg complex, the additional Mg forms three Mg–Si bonds, whereas four Mg-Si bonds are formed in the layered VMg4Si8 + Mg complex. Therefore, the formation and binding energies of layered VMg4Si8 + Mg are lower than those of the most stable VMg4Si8 + Mg complex.
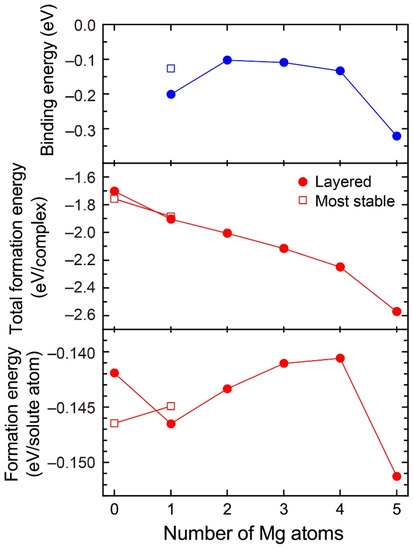
Figure 3.
Binding energy of Mg atom, total formation energy per complex, and formation energy per solute atom for the VMg4Si8 + Mgn complexes as a function of the number of additional Mg atoms.

Figure 4.
Structures of VMg4Si8 + Mgn (n = 1–5) complexes. (a) VMg4Si8 + Mg1 (b) VMg4Si8 + Mg2 (c) VMg4Si8 + Mg3 (d) VMg4Si8 + Mg4 (e) VMg4Si8 + Mg5.
The central Mg atom in the top Mg layer of the VMg4Si8 + Mgn complexes gradually shifted to the Si layer up to n = 4. In the layered VMg4Si8 + Mg5 complex, the central Mg atom completely shifted to the Si layer, and a Mg vacancy was formed in the Mg layer, which indicates that the β″-eye was formed. The formation of the β″-eye was also observed using a larger supercell (shown in Appendix B, Figure A2). Figure 5 shows the average lengths of the Mg–Si, Si–Si, and Mg–Mg bonds in the central Mg and Si layers in the VMg4Si8 + Mgn (n = 1–5) complexes. The average length of the Mg–Si bond remains almost constant at 2.80 Å, which is shorter than that in the Al matrix (2.86 Å). In the VMg4Si8 complex, the Si atoms were outwardly displaced. Therefore, the Si–Si bond is longer than the Mg–Si and the Mg–Mg bonds. The central Mg atom shifted to the Si layer with increasing Mg atoms to form Mg–Si bonds. In addition, the repulsive Mg–Mg interaction around the central Mg increased with the number of Mg atoms, which also contributed to the downward shift of the central Mg atom. The steep decrease in the formation and binding energies of the VMg4Si8 + Mg5 complex arises from not only the formation of Mg–Si bonds but also the weakening of the repulsive Mg–Mg interaction. As a result of the shift of the central Mg atom, the average Si–Si bond length increased from 3.000 to 3.695 Å, which is 0.280 Å shorter than that in the β″-eye in β″-Mg5Si6. This is because the Al matrix suppresses the outward displacement of the Si atoms. The energy loss of the Si–Si bonds is not significant because the contribution of the Si–Si bond to the stability of the VMg4Si8 complexes is smaller than that of the Mg–Si, Al–Mg, and Al–Si bonds [7].
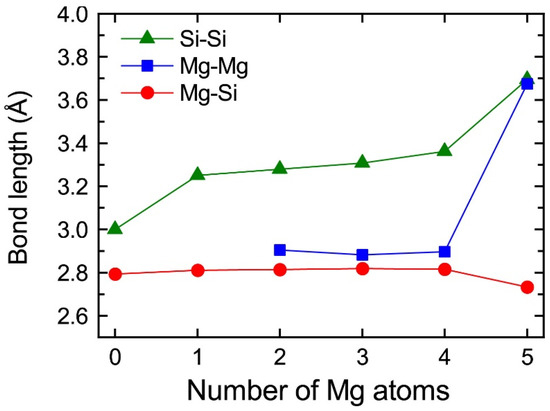
Figure 5.
Average lengths of the Mg–Si, Si–Si, and Mg–Mg bonds in the top Mg and Si layers in VMg4Si8 + Mgn complexes as a function of the number of additional Mg atoms.
To evaluate the change in the stability of the β″-eye with increasing Mg atoms, we calculated the formation energy of the layered VMg4Si8 + Mgn (n = 1–5) complexes with the β″-eye arrangement. The central Mg atom was fixed in the Si layer, which is equivalent to the structure of the β″-eye. The formation energies are shown in Figure 6 with those of the layered VMg4Si8 + Mgn complexes. The formation energy of the β″-eye steeply decreases with increasing Mg atoms and turns out to be lower than that of the layered VMg4Si8 + Mgn complexes for n = 5. The increase in the number of the Mg atoms, which possess the larger atomic radius than the Al atoms, induces the outward displacement of the Si atoms and the Al matrix. Therefore, the Si–Si bonds are expanded as shown in Figure 5, and the structure of the β″-eye becomes stable.
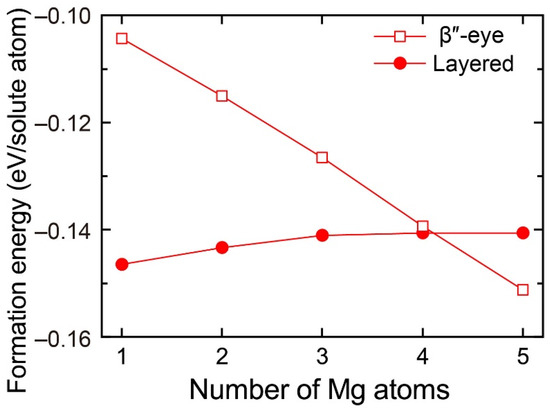
Figure 6.
Formation energies of the layered VMg4Si8 + Mgn complexes with the β″-eye arrangement denoted by open squares as a function of the number of additional Mg atoms. Solid circles denote the formation energies of the layered VMg4Si8 + Mgn complexes in which the central atom is located in the additional Mg layer.
The structure in which the vacancy in VMg4Si8 is replaced by a Mg atom is equivalent to the L10 structure. However, in the L10 structure, the Mg atom does not shift to the adjacent Si layer because the Si–Si bonds are not expanded. Once the β″-eye is formed on the VMg4Si8 complex, the formation of the β″-eye can be repeated by the formation of alternate Mg and Si layers along [10] β″. These results clearly indicate that the layered VMg4Si8 complex plays an important role in the formation of the β″-eye. In the case wherein the first-principles calculation was performed for the VMg4Si8 + Mg5 complex using an initial structure in which the Mg and Si atoms are located on the ideal FCC lattice sites, the Mg atom in the top Mg layer does not completely shift to the Si layer. This is because lattice relaxation is confined by symmetry restriction. Therefore, the atom-by-atom formation of the Mg layer is also important for the β″-eye formation on the VMg4Si8 + Mg5 complex.
To reproduce the formation of the β″-eye from the solid solution in the Al–Mg–Si alloy, we performed the first-principles-based MC simulations using a supercell composed of 108 lattice sites with an FCC structure, including a vacancy and Mg and Si atoms. Figure 7 shows the change in the potential energy of Al83Mg12Si12, in which the β″-eye was formed during the MC simulation. For comparison, we performed MC simulations for Al84Mg12Si12, in which a vacancy was not included. The β″-eye was formed at 68 MC steps in Al83Mg12Si12. Figure 8 shows the structures before and after the β″-eye formation. While the vacancy–solute complex that appeared in Al83Mg12Si12 is not VMg4Si8 but VMg4Si7, the central Mg atom in the top Mg layer completely shifts to the Si layer. In contrast, the structure of Al84Mg12Si12, in which a vacancy is not included, exhibits an L10-like layered structure, which agrees with the result of a multiscale approach [15]. If a Mg vacancy is formed in the L10 structure, the local structure around the Mg vacancy becomes similar to that of the layered VMg4Si8 complex. To examine the possibility of β″-eye formation around the Mg vacancy in the L10 structure, we investigated the change in the structure upon the addition of Mg atoms for the layered MgSi with a Mg vacancy. Figure 9 shows the structures in which up to five Mg atoms were located in the Si layer above the Mg vacancy. Although the central Mg atom shifted to the Si layer with increasing Mg atoms, the central Mg atom still existed between the Mg and Si layers. This is partly because the Si–Si bond was not well expanded to accommodate the Mg–Si bonds in the Si layer. The local structure around the Mg vacancies may be affected by the composition or size of the MgSi layers. Therefore, further research is necessary to clarify the possibility of the formation of the β″-eye on the MgSi layers. Note that the β″-eye was not formed on layered MgSi with a Mg vacancy in the present work.

Figure 7.
Change in the potential energy of Al83Mg12Si12 with a vacancy and Al84Mg12Si12 during the MC simulation.

Figure 8.
Structures (a) before and (b) after the β″-eye was formed during the MC simulation in Al83Mg12Si12 with a vacancy. (c) Structure of Al84Mg12Si12 after the MC simulation.

Figure 9.
Structures of the layered MgSi including a Mg vacancy with additional Mg atoms on the Si layer. (a) MgSi-VMg+Mg1 (b) MgSi-VMg+Mg2 (c) MgSi-VMg+Mg3 (d) MgSi-VMg+Mg4 (e) MgSi-VMg+Mg5
The layered VMg4Si8 + Mgn complex appeared in other compositions of the MC simulations, as shown in Figure 10. In Al89Mg6Si12 and Al89Mg9Si9, the layered VMg4Si8 + Mg2 and VMg4Si8 + Mg5 complexes were formed, respectively; in these complexes, the number of Mg atoms in the additional Mg layer was not sufficient to form the β″-eye. In the MC simulations performed in this study, the most stable VMg4Si8 complex was not observed, which suggests that the structural transition from the most stable VMg4Si8 complex is difficult to proceed. At lower temperatures, the amount of the layered VMg4Si8 complex decreases compared with that of the most stable VMg4Si8 complex, which is considered one of the reasons for the decrease in the peak hardness value obtained by the precipitation of the β″ phase by natural aging. As seen in the results of the MC simulations, more than 10 Mg atoms are required to reproduce the β″-eye formation. Therefore, to quantitatively evaluate the influence of Mg/Si ratios in the low concentration region on β″-eye formation efficiency, a larger supercell must be employed.

Figure 10.
Structures of (a) Al89Mg6Si12 with a vacancy and (b) Al89Mg9Si9 with a vacancy after the MC simulation.
4. Conclusions
We investigated the stability and structure of vacancy–solute complexes VMg4Si8 with additional Mg atoms in Al–Mg–Si alloys using the first-principles calculations. The layered VMg4Si8 complex, which is the second most stable complex of VMg4Si8, becomes stable upon the addition of Mg atoms to the adjacent layers because of the formation of Mg–Si bonds. The central Mg atom in the additional Mg layer shifted to the Si layer with the increase in the number of Mg atoms to weaken the repulsive Mg–Mg interaction as well as to form Mg–Si bonds. When five Mg atoms were added to the layered VMg4Si8 complex, the central Mg atom completely shifted to the Si layer, and a Mg vacancy is formed in the Mg layer. Therefore, the layered VMg4Si8 + Mg5 complex has the same structure as the β″-eye. These results indicate that the layered VMg4Si8 complex evolves into the β″-eye upon the addition of Mg atoms. In contrast, the layered MgSi with a Mg vacancy, which has a similar structure to the layered VMg4Si8 complex, does not evolve into the β″-eye. We confirmed the formation of the β″-eye from a solid solution with a vacancy using first-principles-based MC simulations. The β″ structure is composed of β″-eyes linked by Si–Si bonds. Therefore, to investigate the evolution from the β″-eye to the β″ phase, larger supercells are required in order to include more than one β″-eye, which will be examined in future work.
Author Contributions
Conceptualization, M.M.; methodology, M.M.; software, M.M.; investigation, M.M. and K.S.; writing—original draft preparation, M.M.; writing—review and editing, M.M., K.S. and H.A.; project administration, H.A.; funding acquisition, M.M. and H.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by a Grant-in-Aid for Scientific Research (C) (grant number 20K05131) from the Japan Society for the Promotion of Science (JSPS), and a research grant from the Light Metal Educational Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
To evaluate the influence of the temperature, we performed the MC simulations for the solid solution of Al95Mg4Si8 with a vacancy at 200 K and 300 K. Figure A1 shows the change in the potential energy. The potential energy at 200 K shows a decreasing trend during the MC simulations. However, the potential energy at 300 K steeply increases between 100 and 120 MC steps and fluctuates around −0.8 eV after 120 MC steps, which suggests that the MC simulations at 300 K are not suitable to find stable structures. This result does not indicate that the heat treatment at 200 K is more suitable for the precipitation of the β″phase because the difference in the atomic jump frequencies of the solute atoms by temperatures is not considered in the MC simulations. The purpose of the MC simulations is not to evaluate the influence of finite temperature but to reproduce the formation of the β″-eye from the solid solution. Therefore, we employed 200 K for the MC simulations in the present work.

Figure A1.
Change in the potential energy of Al95Mg4Si8 during the MC simulation at 200 K and 300 K.
Figure A1.
Change in the potential energy of Al95Mg4Si8 during the MC simulation at 200 K and 300 K.

Appendix B
In the 3 × 3 × 3 supercell including the layered VMg4Si8 complex, there are three Al matrix layers between the layered VMg4Si8 complex. In the case in which the layered VMg4Si8 + Mgn complex is included in the 3 × 3 × 3 supercell, the number of the Al matrix layers along the stacking direction of the additional Mg layer decreases to two layers. To evaluate the influence of the Al matrix layers, the formation energies of VMg4Si8 + Mgn (n = 1–5) complexes were calculated using the 3 × 3 × 4 supercell, which was expanded along the stacking direction of the additional Mg layer. Figure A2 shows the formation energies of VMg4Si8 + Mgn (n = 1–5) complexes calculated using the 3 × 3 × 4 supercell and the distance of the central Mg atom in the additional Mg layer from the Si layer. For comparison, the calculated results obtained using the 3 × 3 × 3 supercell are shown in Figure A2. While the formation energies calculated with the 3 × 3 × 3 supercell tend to be lower, the changes in the formation energies from increasing the number of Mg atoms are very similar between the 3 × 3 × 3 and 3 × 3 × 4 supercells. In addition, there is no significant difference in the shift of the central Mg atom to the Si layer, which is the most important feature in the present work, between the two supercells. Therefore, the increase in the supercell size does not affect the conclusions of the present work.

Figure A2.
Formation energies of VMg4Si8 + Mgn complexes calculated using the 3 × 3 × 3 and 3 × 3 × 4 supercells and the distance of the central Mg atom in the additional Mg layer from the Si layer.
Figure A2.
Formation energies of VMg4Si8 + Mgn complexes calculated using the 3 × 3 × 3 and 3 × 3 × 4 supercells and the distance of the central Mg atom in the additional Mg layer from the Si layer.

References
- Andersen, S.J.; Zandbergen, H.W.; Jansen, J.; TrÆholt, C.; Tundal, U.; Reiso, O. The crystal structure of the β″ phase in Al–Mg–Si alloys. Acta Mater. 1998, 46, 3283–3298. [Google Scholar] [CrossRef]
- Marioara, C.D.; Andersen, S.J.; Jansen, J.; Zandbergen, H.W. Atomic model for GP-zones in a 6082 Al–Mg–Si system. Acta Mater. 2001, 49, 321–328. [Google Scholar] [CrossRef]
- Marioara, C.D.; Andersen, S.J.; Jansen, J.; Zandbergen, H.W. The influence of temperature and storage time at RT on nucleation of the β″ phase in a 6082 Al–Mg–Si alloy. Acta Mater. 2003, 51, 789–796. [Google Scholar] [CrossRef]
- Serizawa, A.; Hirosawa, S.; Sato, T. Three-dimensional atom probe characterization of nanoclusters responsible for multistep aging behavior of an Al-Mg-Si alloy. Metall. Mater. Trans. A 2008, 39, 243–251. [Google Scholar] [CrossRef]
- Ding, L.; Jia, Z.; Liu, Y.; Weng, Y.; Liu, Q. The influence of Cu addition and pre-straining on the natural aging and bake hardening response of Al-Mg-Si alloys. J. Alloys Compd. 2016, 688, 362–367. [Google Scholar] [CrossRef]
- Ding, L.; Weng, Y.; Wu, S.; Sanders, R.E.; Jia, Z.; Liu, Q. Influence of interrupted quenching and pre-aging on the bake hardening of Al-Mg-Si alloy. Mater. Sci. Eng. A 2016, 651, 991–998. [Google Scholar] [CrossRef]
- Mizuno, M.; Sugita, K.; Araki, H. Structure and stability of vacancy–solute complexes in Al–Mg–Si alloys. Materialia 2020, 13, 100853. [Google Scholar] [CrossRef]
- Derlet, P.M.; Andersen, S.J.; Marioara, C.D.; Frøseth, A. A first-principles study of the βʺ-phase in Al–Mg–Si alloys. J. Phys. Condens. Matter 2002, 14, 4011–4024. [Google Scholar]
- Frøseth, A.G.; Høier, R.; Derlet, P.M.; Andersen, S.J.; Marioara, C.D. Bonding in MgSi and Al-Mg-Si compounds relevant to Al-Mg-Si alloys. Phys. Rev. B 2003, 67, 224106. [Google Scholar] [CrossRef]
- Ravi, C.; Wolverton, C. First-principles study of crystal structure and stability of Al–Mg–Si−(Cu) precipitates. Acta Mater. 2004, 52, 4213–4227. [Google Scholar] [CrossRef]
- van Huis, M.A.; Chen, J.H.; Sluiter, M.H.F.; Zandbergen, H.W. Phase stability and structural features of matrix-embedded hardening precipitates in Al–Mg–Si alloys in the early stages of evolution. Acta Mater. 2007, 55, 2183–2199. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.-K.; Chen, L.-Q.; Wolverton, C. First-principles calculations of β″-Mg5Si6/α-Al interfaces of βʺ-Mg5Si6/α-Al interfaces. Acta Mater. 2007, 55, 5934–5947. [Google Scholar] [CrossRef]
- Hasting, H.S.; Frøseth, A.G.; Andersen, S.J.; Vissers, R.; Walmsley, J.C.; Marioara, C.D.; Danoix, F.; Lefebvre, W.; Holmestad, R. Composition of β″ precipitates in Al–Mg–Si alloys by atom probe tomography and first principles calculations. J. Appl. Phys. 2009, 106, 123527. [Google Scholar] [CrossRef]
- Huan, T.D.; Le, N.B. Characterizing magnesium silicon binaries in Al–Mg–Si supersaturated solid solution by first-principles calculations. J. Sci. Adv. Mater. Dev. 2016, 1, 527–530. [Google Scholar] [CrossRef][Green Version]
- Kleiven, D.; Akola, J. Precipitate formation in aluminium alloys: Multi-scale modelling approach. Acta Mater. 2020, 195, 123–131. [Google Scholar] [CrossRef]
- Zandbergen, H.W.; Andersen, S.; Jansen, J. Structure Determination of Mg5Si6 particles in Al by dynamic electron diffraction studies. Science 1997, 227, 1221–1225. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).