
Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method


Abstract
1. Introduction
2. Calculation Methods
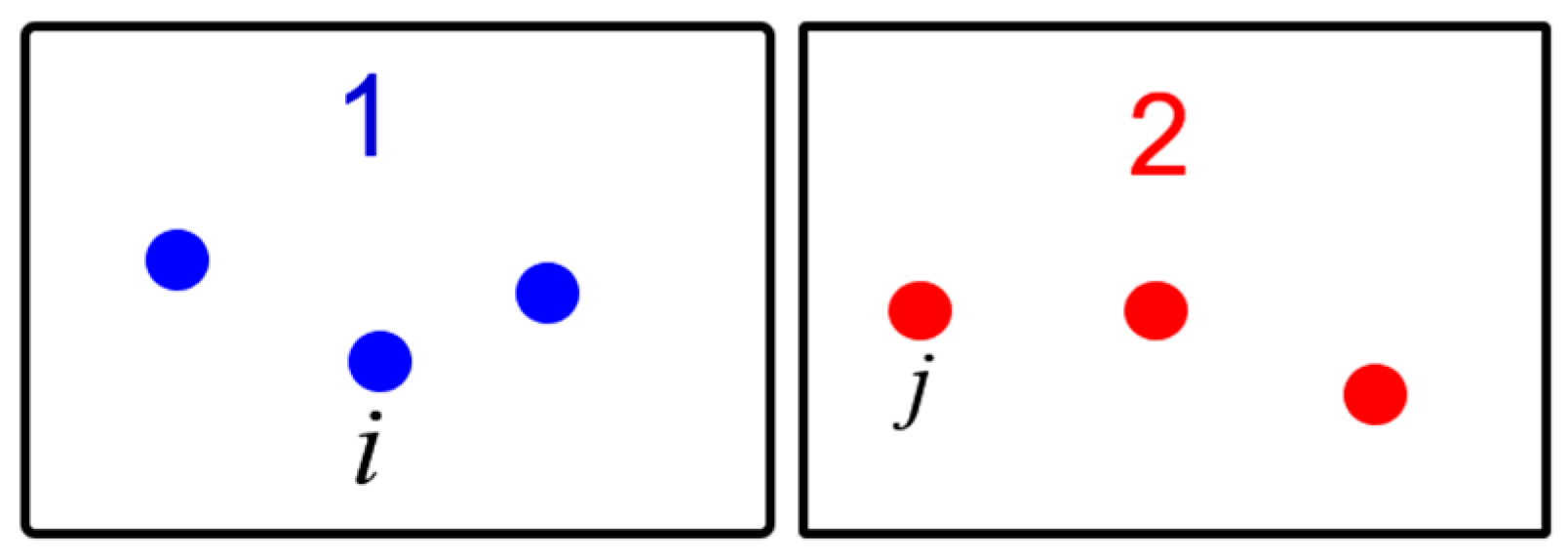
2.1. Lattice Inversion
2.2. Interface Thermal Conductance
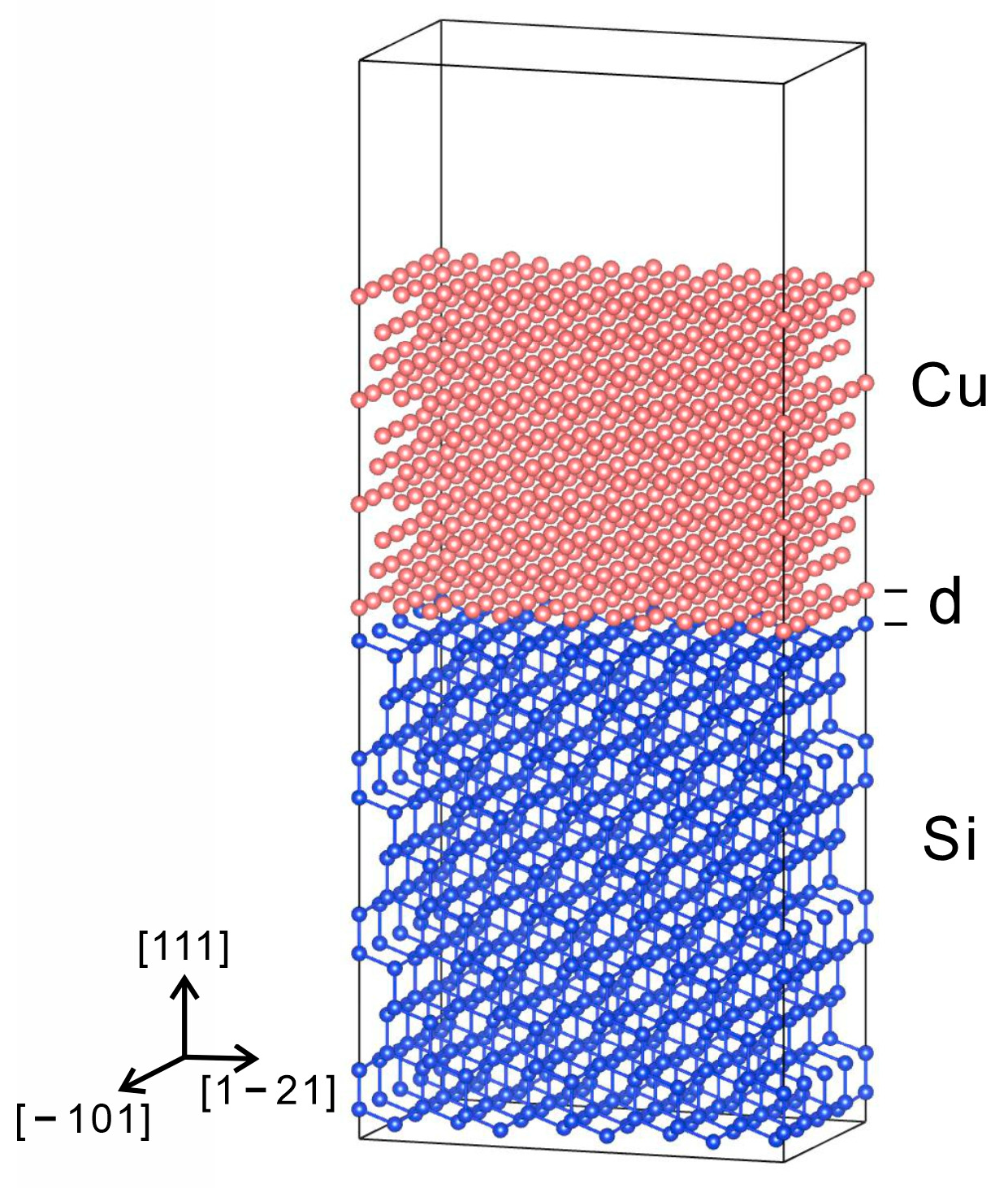
2.3. Computational Details
3. Results and Discussions
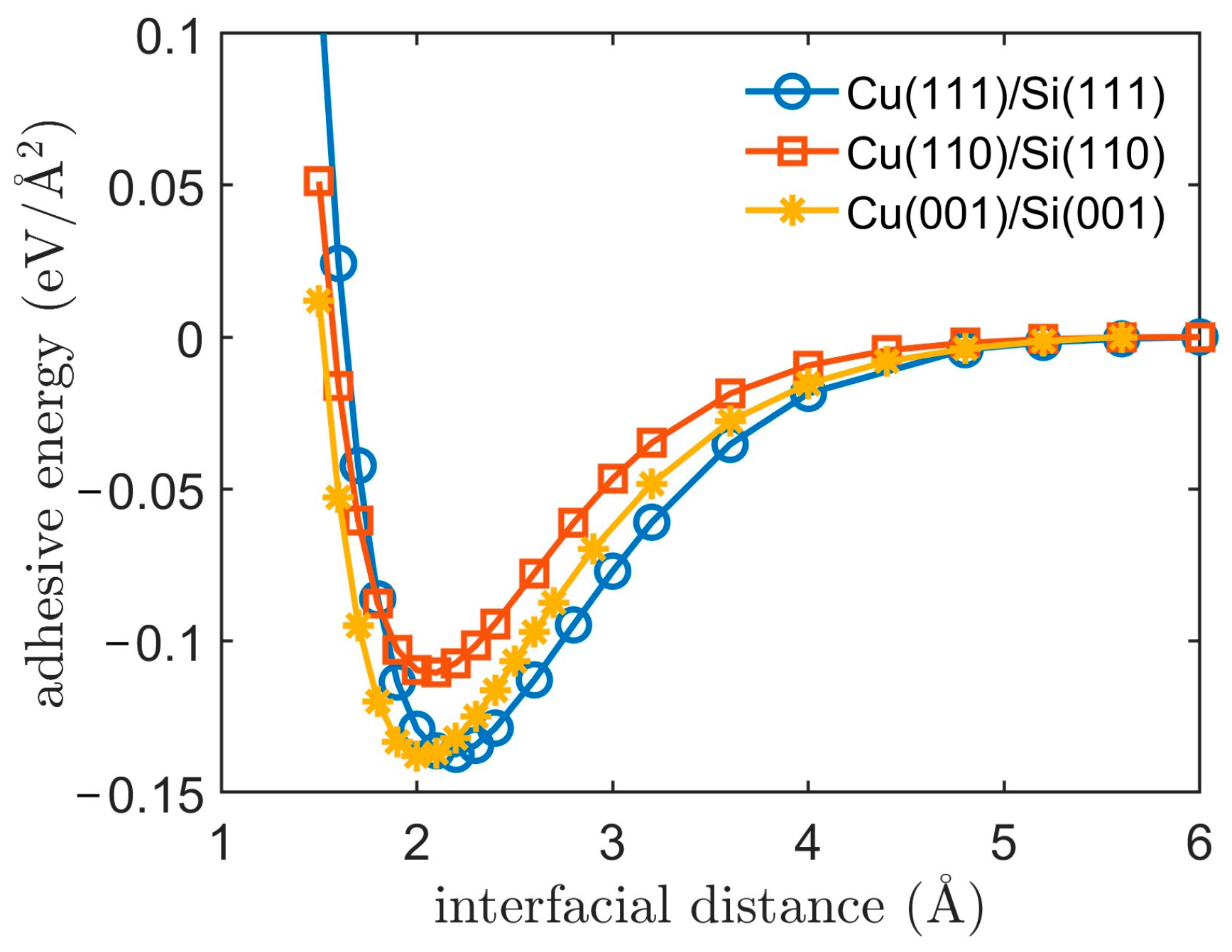
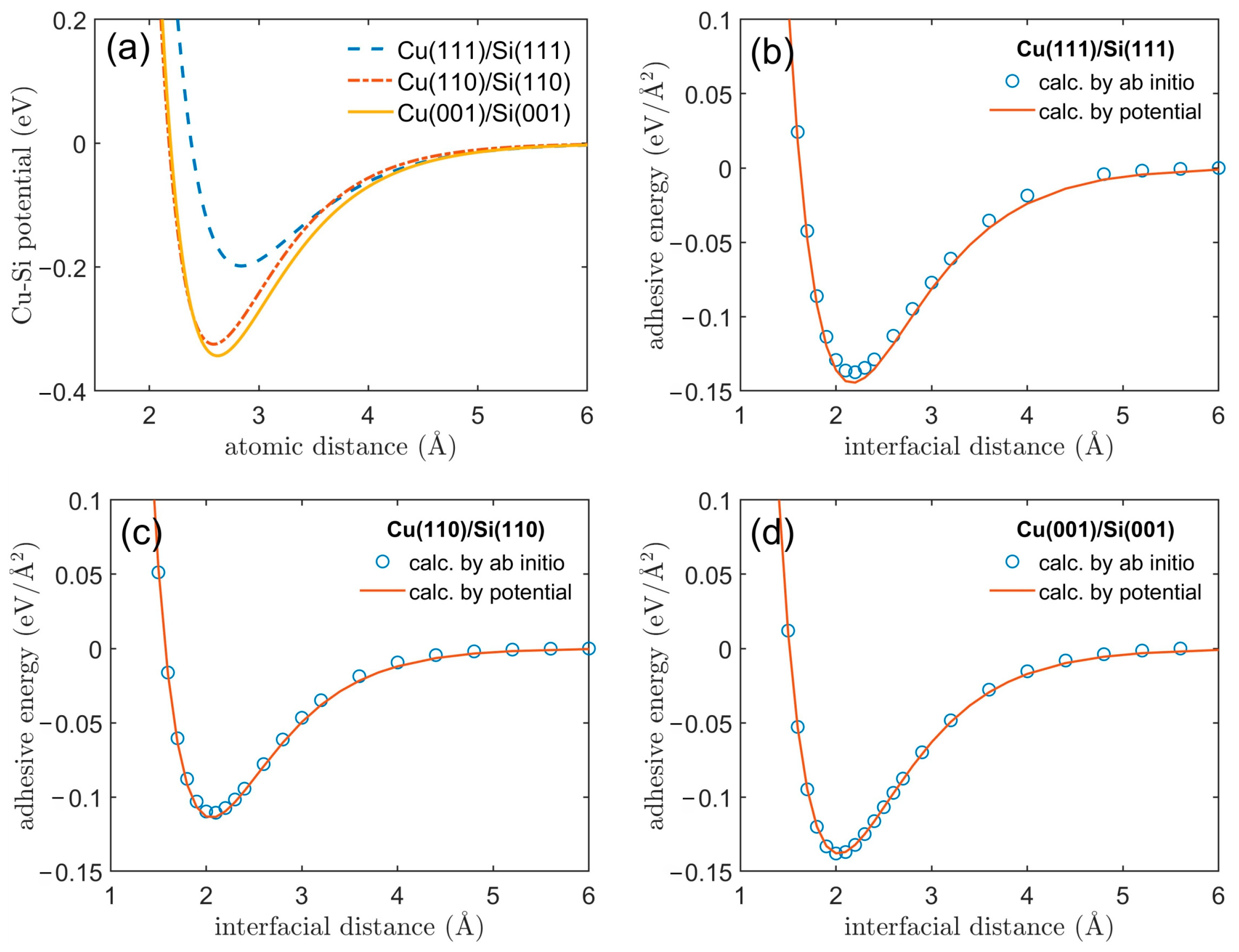
3.1. Adhesive Energy

3.2. Cu/Si Potential

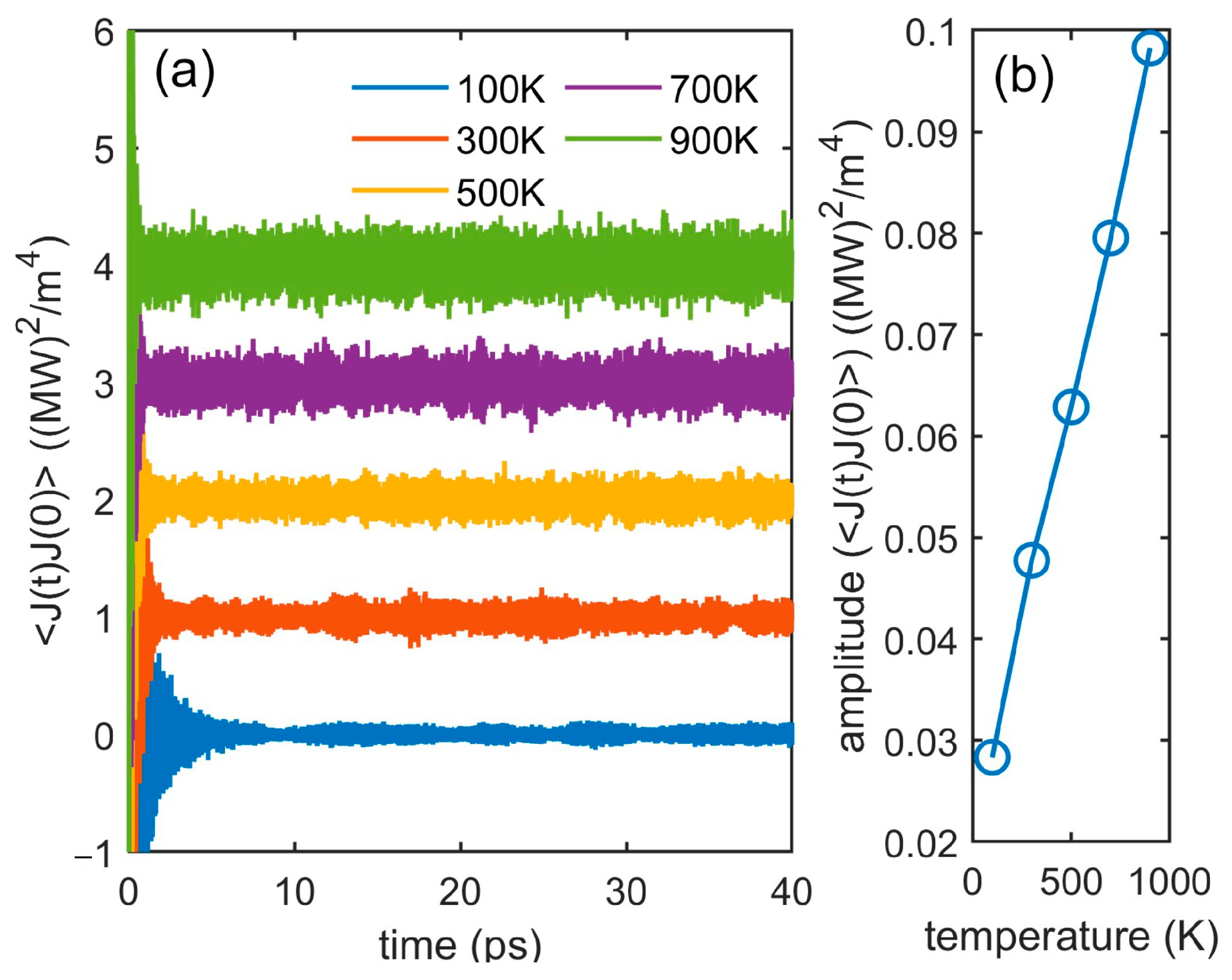
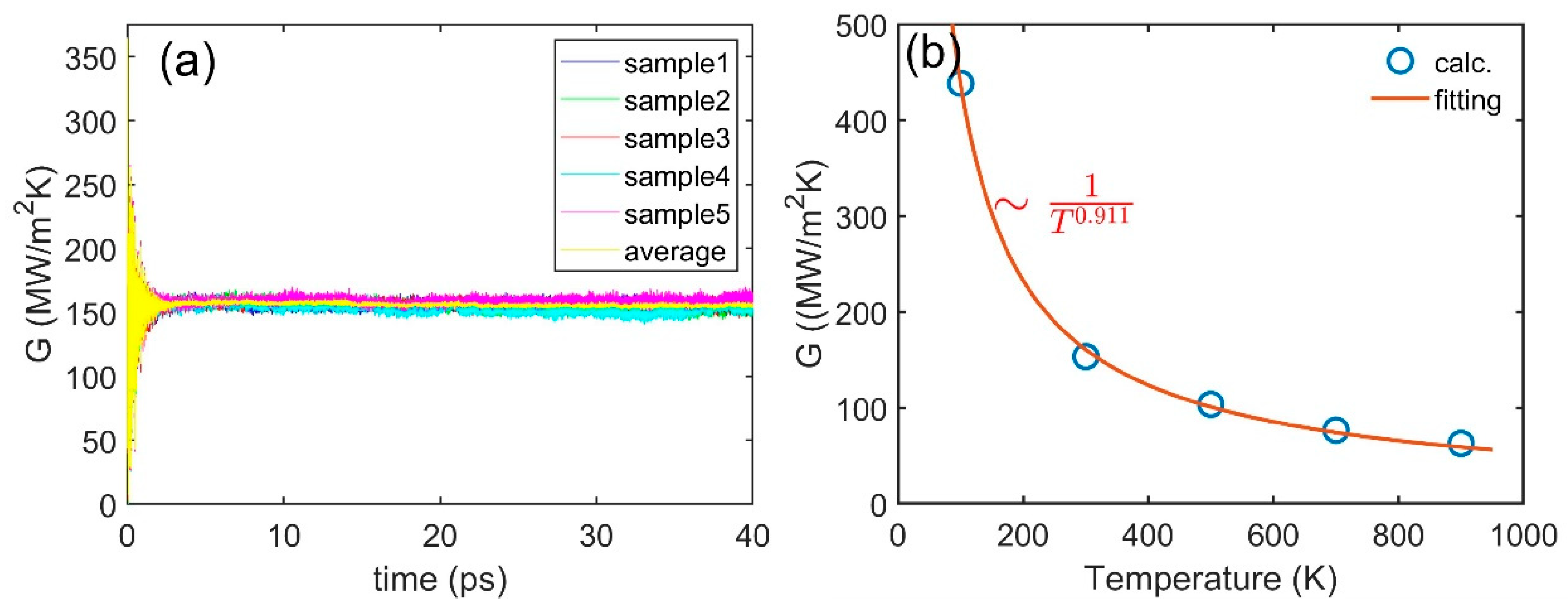
3.3. Interface Thermal Conductance
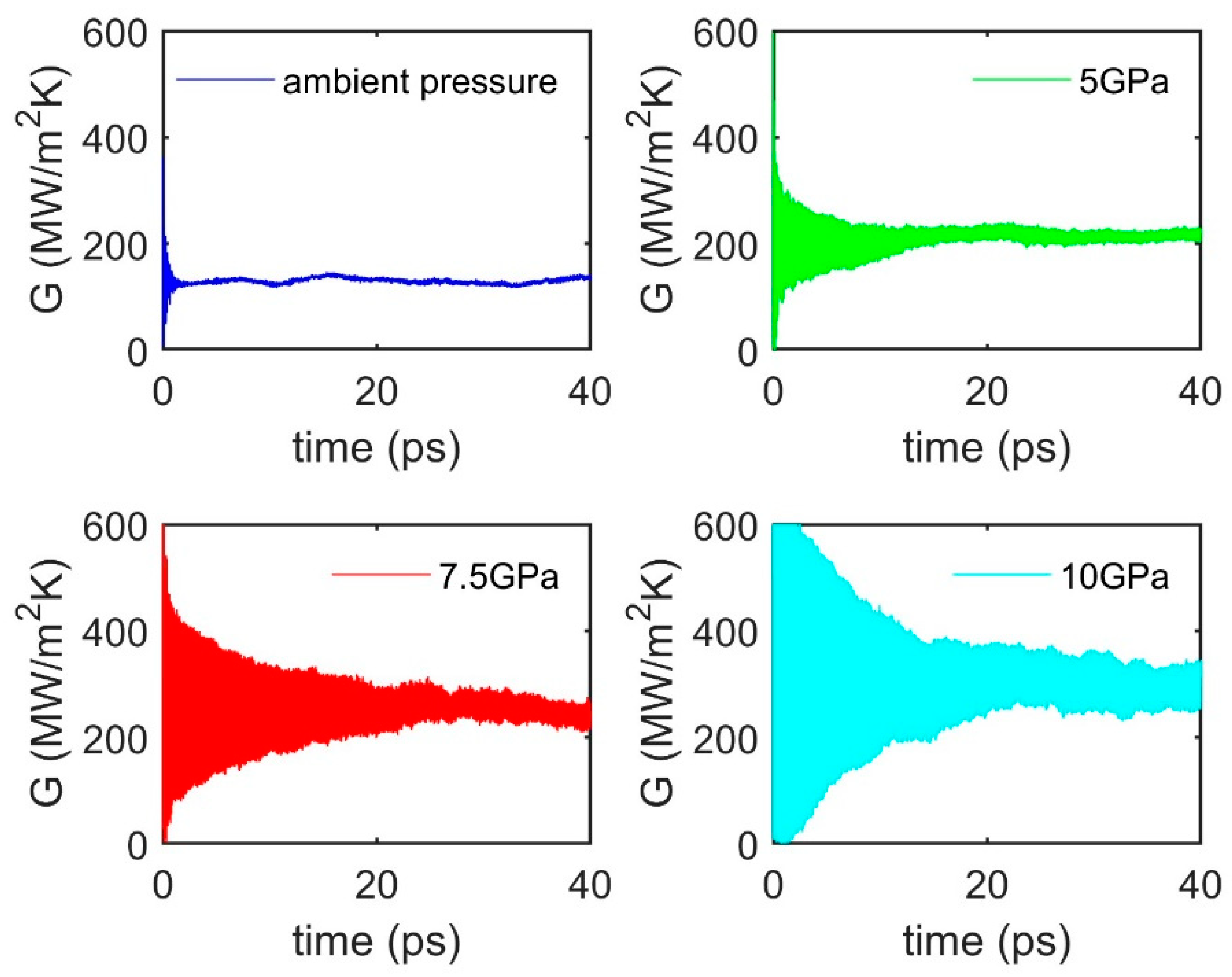
3.4. Pressure Effect
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, W.; Li, T.; Tong, Y.; Li, Y.; Wang, H.; Qi, H.; Wang, K.; Wang, H. Reducing Nonradiative Losses of Air-Processed Perovskite Films via Interface Modification for Bright and Efficient Light Emitting Diodes. Adv. Funct. Mater. 2024, 34, 2311133. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, L.; Yan, M.; Luo, G.; Yang, D.; Fang, Y. High-Brightness Thermally Evaporated Perovskite Light-Emitting Diodes via Dual-Interface Engineering. Opt. Mater. 2024, 150, 115223. [Google Scholar] [CrossRef]
- Lee, W.J.; Sohn, W.B.; Shin, J.C.; Han, I.K.; Kim, T.G.; Kang, J. Growth of InGaAs/InAlAs Superlattices for Strain Balanced Quantum Cascade Lasers by Molecular Beam Epitaxy. J. Cryst. Growth 2023, 614, 127233. [Google Scholar] [CrossRef]
- Knipfer, B.; Xu, S.; Kirch, J.D.; Botez, D.; Mawst, L.J. Analysis of Interface Roughness in Strained InGaAs/AlInAs Quantum Cascade Laser Structures (λ∼4.6 μm) by Atom Probe Tomography. J. Cryst. Growth 2022, 583, 126531. [Google Scholar] [CrossRef]
- Khan, A.I.; Wu, X.; Perez, C.; Won, B.; Kim, K.; Ramesh, P.; Kwon, H.; Tung, M.C.; Lee, Z.; Oh, I.-K.; et al. Unveiling the Effect of Superlattice Interfaces and Intermixing on Phase Change Memory Performance. Nano Lett. 2022, 22, 6285–6291. [Google Scholar] [CrossRef] [PubMed]
- Aryana, K.; Gaskins, J.T.; Nag, J.; Stewart, D.A.; Bai, Z.; Mukhopadhyay, S.; Read, J.C.; Olson, D.H.; Hoglund, E.R.; Howe, J.M.; et al. Interface Controlled Thermal Resistances of Ultra-Thin Chalcogenide-Based Phase Change Memory Devices. Nat. Commun. 2021, 12, 774. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Park, S.J.; Kim, G.-Y.; Choi, S.-Y.; Jin, H. Designing Efficient Spin Seebeck-Based Thermoelectric Devices via Simultaneous Optimization of Bulk and Interface Properties. Energy Environ. Sci. 2021, 14, 3480–3491. [Google Scholar] [CrossRef]
- Wu, W.; Ren, G.-K.; Chen, X.; Liu, Y.; Zhou, Z.; Song, J.; Shi, Y.; Jiang, J.-M.; Lin, Y.-H. Interfacial Advances Yielding High Efficiencies for Thermoelectric Devices. J. Mater. Chem. A 2021, 9, 3209–3230. [Google Scholar] [CrossRef]
- Kim, H.; Kwon, Y.; Lim, H.; Kim, J.; Kim, Y.; Yeo, W. Recent Advances in Wearable Sensors and Integrated Functional Devices for Virtual and Augmented Reality Applications. Adv. Funct. Mater. 2021, 31, 2005692. [Google Scholar] [CrossRef]
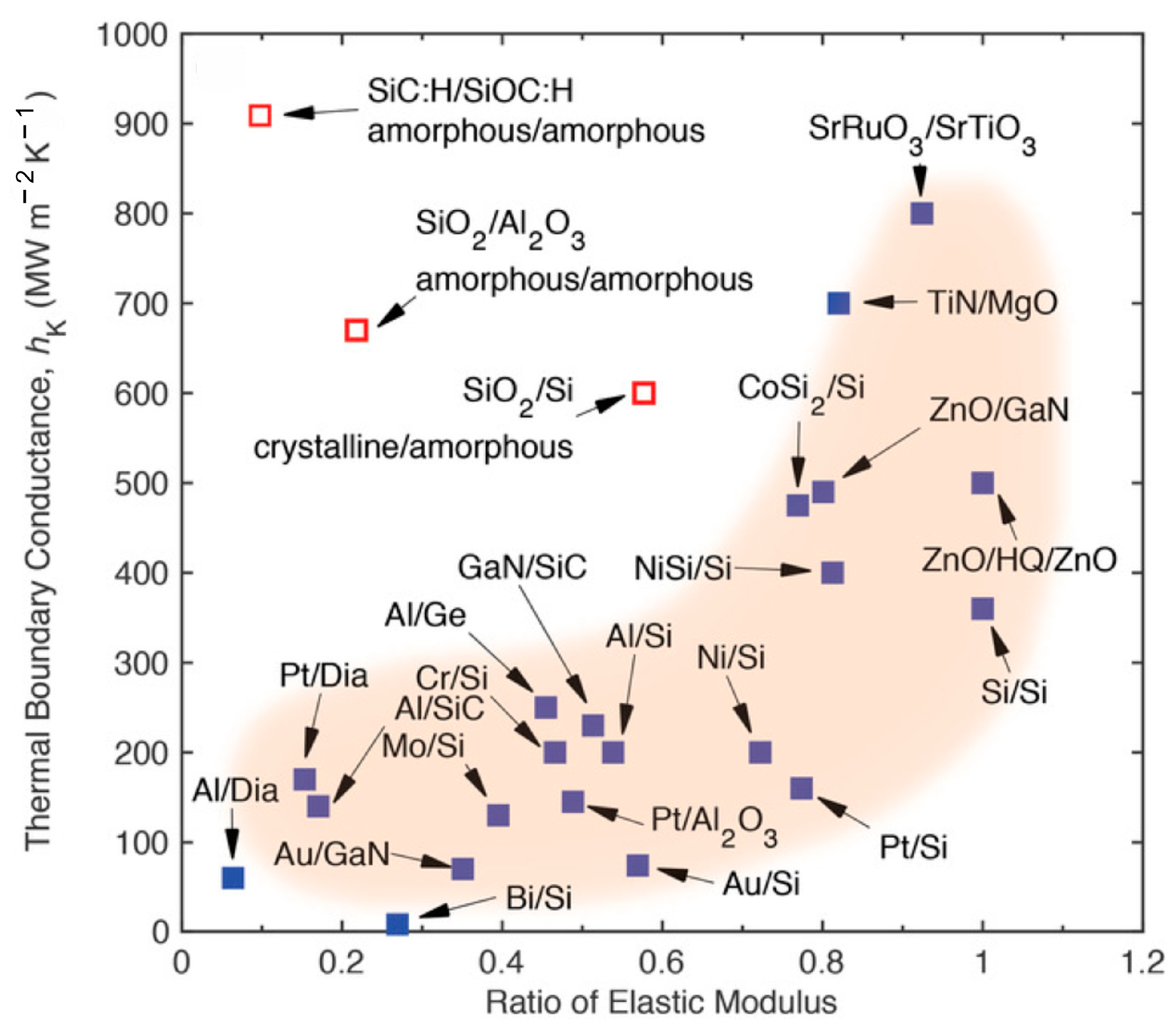
- Giri, A.; Hopkins, P.E. A Review of Experimental and Computational Advances in Thermal Boundary Conductance and Nanoscale Thermal Transport across Solid Interfaces. Adv. Funct. Mater. 2020, 30, 1903857. [Google Scholar] [CrossRef]
- Findik, F. Laser Cladding and Applications. Sustain. Eng. Innov. 2023, 5, 1–14. [Google Scholar] [CrossRef]
- Chen, J. Interfacial Thermal Resistance: Past, Present, and Future. Rev. Mod. Phys. 2022, 94, 25002. [Google Scholar] [CrossRef]
- Swartz, E.T.; Pohl, R.O. Thermal Boundary Resistance. Rev. Mod. Phys. 1989, 61, 605–668. [Google Scholar] [CrossRef]
- Liang, Z.; Hu, M. Tutorial: Determination of Thermal Boundary Resistance by Molecular Dynamics Simulations. J. Appl. Phys. 2018, 123, 191101. [Google Scholar] [CrossRef]
- Little, W.A. The transport of heat between dissimilar solids at low temperatures. Can. J. Phys. 1959, 37, 334–349. [Google Scholar] [CrossRef]
- Li, Q.; Liu, F.; Hu, S.; Song, H.; Yang, S.; Jiang, H.; Wang, T.; Koh, Y.K.; Zhao, C.; Kang, F.; et al. Inelastic Phonon Transport across Atomically Sharp Metal/Semiconductor Interfaces. Nat. Commun. 2022, 13, 4901. [Google Scholar] [CrossRef] [PubMed]
- Cheaito, R.; Gaskins, J.T.; Caplan, M.E.; Donovan, B.F.; Foley, B.M.; Giri, A.; Duda, J.C.; Szwejkowski, C.J.; Constantin, C.; Brown-Shaklee, H.J.; et al. Thermal Boundary Conductance Accumulation and Interfacial Phonon Transmission: Measurements and Theory. Phys. Rev. B—Condens. Matter Mater. Phys. 2015, 91, 035432. [Google Scholar] [CrossRef]
- Stevens, R.J.; Smith, A.N.; Norris, P.M. Measurement of Thermal Boundary Conductance of a Series of Metal-Dielectric Interfaces by the Transient Thermoreflectance Technique. J. Heat Transfer 2005, 127, 315–322. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, Y.; Ruan, X. Metal/Dielectric Thermal Interfacial Transport Considering Cross-Interface Electron-Phonon Coupling: Theory, Two-Temperature Molecular Dynamics, and Thermal Circuit. Phys. Rev. B 2016, 93, 064302. [Google Scholar] [CrossRef]
- Abs da Cruz, C.; Chantrenne, P.; Gomes de Aguiar Veiga, R.; Perez, M.; Kleber, X. Modified Embedded-Atom Method Interatomic Potential and Interfacial Thermal Conductance of Si-Cu Systems: A Molecular Dynamics Study. J. Appl. Phys. 2013, 113, 023710. [Google Scholar] [CrossRef]
- Chen, N. Modified Möbius Inverse Formula and Its Applications in Physics. Phys. Rev. Lett. 1990, 64, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Chen, N.X.; Zhang, W.Q. Pair Potentials for a Metal–Ceramic Interface by Inversion of Adhesive Energy. J. Phys. Condens. Matter 2005, 17, 2045–2058. [Google Scholar] [CrossRef]
- Long, Y.; Chen, N.X. Pair Potential Approach for Metal/Al2O3 Interface. J. Phys. Condens. Matter 2007, 19, 196216. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, N.; Long, Y. Interfacial Potentials for Al/SiC(111). J. Phys. Condens. Matter 2009, 21, 225002. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Q.; Song, H.-Q.; Shen, J. Interfacial Potential Approach for Ag/Si(111) Interface. Mod. Phys. Lett. B 2016, 30, 1650104. [Google Scholar] [CrossRef]
- Domingues, G.; Volz, S.; Joulain, K.; Greffet, J. Heat Transfer between Two Nanoparticles Through Near Field Interaction. Phys. Rev. Lett. 2005, 94, 085901. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Johnson, R.A. Alloy Models with the Embedded-Atom Method. Phys. Rev. B 1989, 39, 12554–12559. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.W.; Johnson, R.A.; Wadley, H.N.G. Misfit-Energy-Increasing Dislocations in Vapor-Deposited CoFe/NiFe Multilayers. Phys. Rev. B 2004, 69, 144113. [Google Scholar] [CrossRef]
- Tersoff, J. Modeling Solid-State Chemistry: Interatomic Potentials for Multicomponent Systems. Phys. Rev. B 1989, 39, 5566–5568. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface Model | D0 (eV) | α | r0 (Å) |
|---|---|---|---|
| Cu(111)/Si(111) | 0.1983 | 1.5188 | 2.8395 |
| Cu(110)/Si(110) | 0.3247 | 1.7009 | 2.5873 |
| Cu(001)/Si(001) | 0.3434 | 1.6154 | 2.6225 |
| Cu/Si [19] | 0.9 | 1.11 | 3.15 |
| Interface Model | Temperature (K) | Pressure (GPa) | G (MW/m2K) |
|---|---|---|---|
| Cu/Si [19] | 300 | - | 436 |
| Cu/Si [20] | 300 | - | 234–263 |
| Cu(110)/Si(110) | 300 | 0 | 113 |
| Cu(001)/Si(001) | 300 | 0 | 192 |
| Cu(111)/Si(111) | 100 | 0 | 438 |
| 300 | 0 | 153 | |
| 500 | 0 | 101 | |
| 700 | 0 | 74 | |
| 900 | 0 | 59 | |
| 300 | Ambient | 125 | |
| 300 | 5 | 213 | |
| 300 | 7.5 | 248 | |
| 300 | 10 | 290 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Zhi, Y.; Song, H.; Li, H.; Wang, W.; Hu, X.; Zhang, D. Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method. Metals 2024, 14, 453. https://doi.org/10.3390/met14040453
Liu S, Zhi Y, Song H, Li H, Wang W, Hu X, Zhang D. Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method. Metals. 2024; 14(4):453. https://doi.org/10.3390/met14040453
Chicago/Turabian StyleLiu, Shuai, Yueyi Zhi, Hongquan Song, Huijin Li, Weiping Wang, Xiaoyan Hu, and Dongbo Zhang. 2024. "Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method" Metals 14, no. 4: 453. https://doi.org/10.3390/met14040453
APA StyleLiu, S., Zhi, Y., Song, H., Li, H., Wang, W., Hu, X., & Zhang, D. (2024). Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method. Metals, 14(4), 453. https://doi.org/10.3390/met14040453