
A Review of Molecular Dynamics Simulation of Different Ti-Al-Based Alloys




Abstract
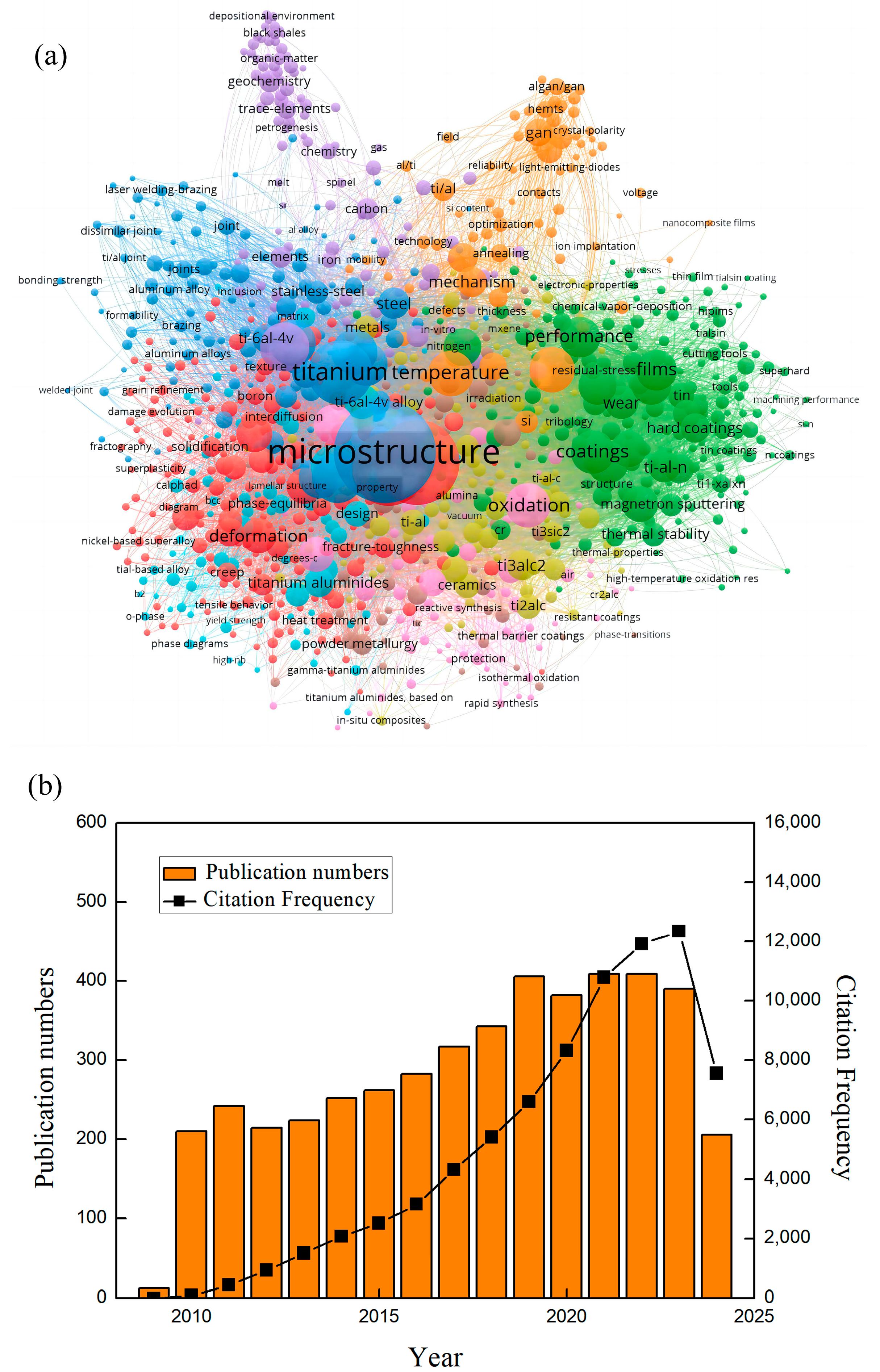
1. Introduction
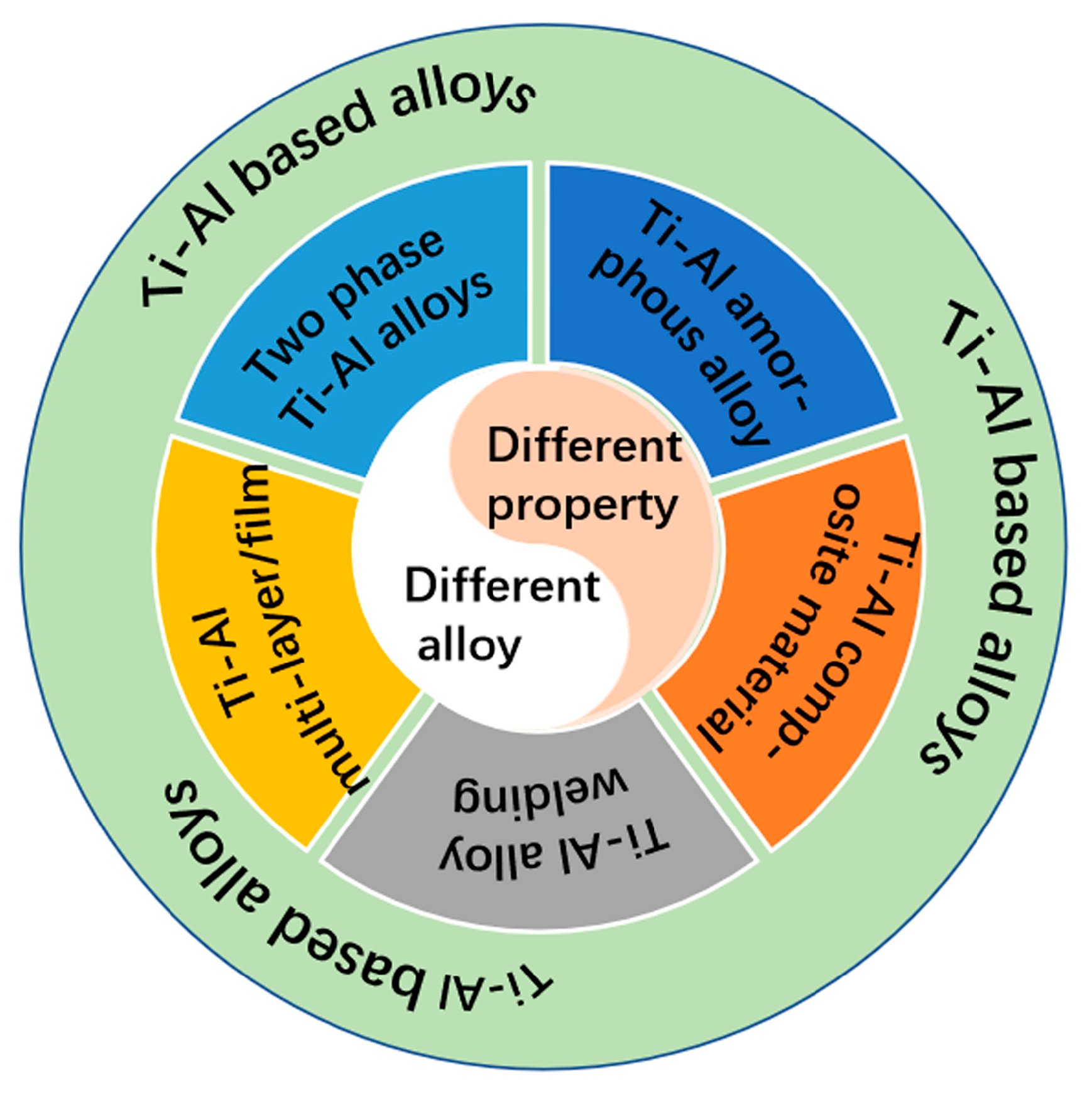
2. Prediction of Different Ti-Al-Based Alloys
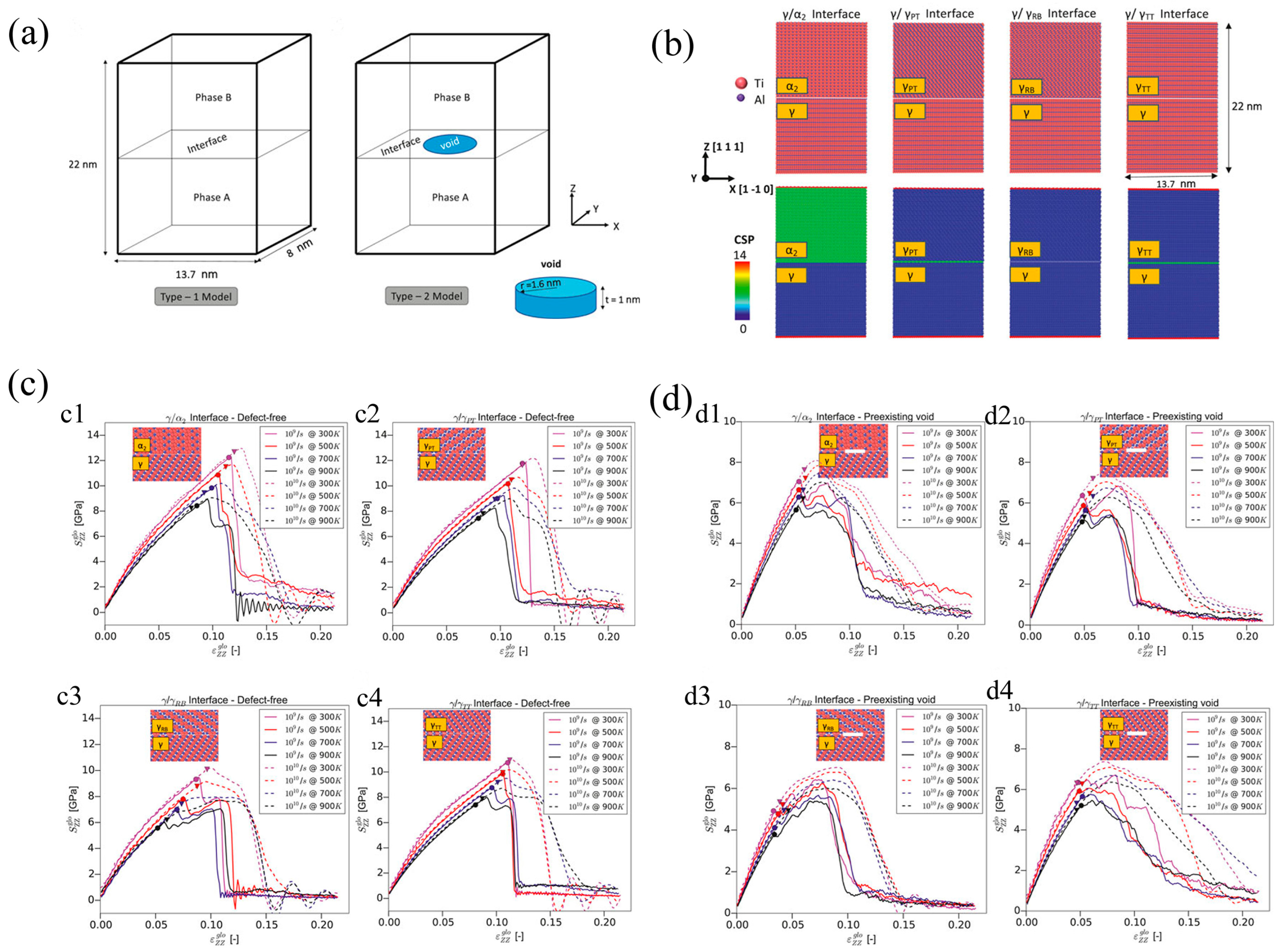
2.1. Simulation of Two-Phase Ti-Al Alloys
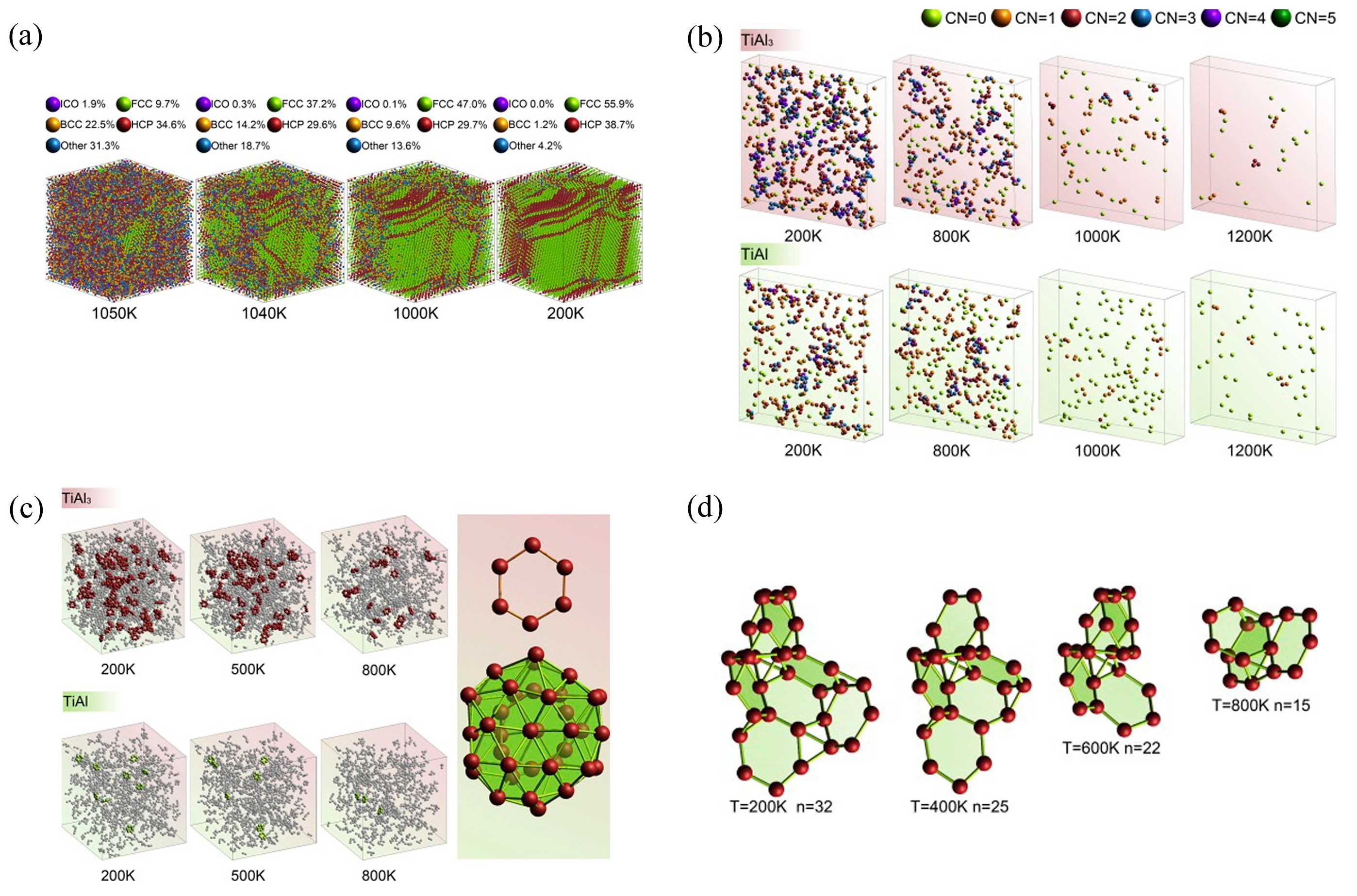
2.2. Simulation of Ti-Al Amorphous Alloys
2.3. Simulation of Ti-Al Composite Material
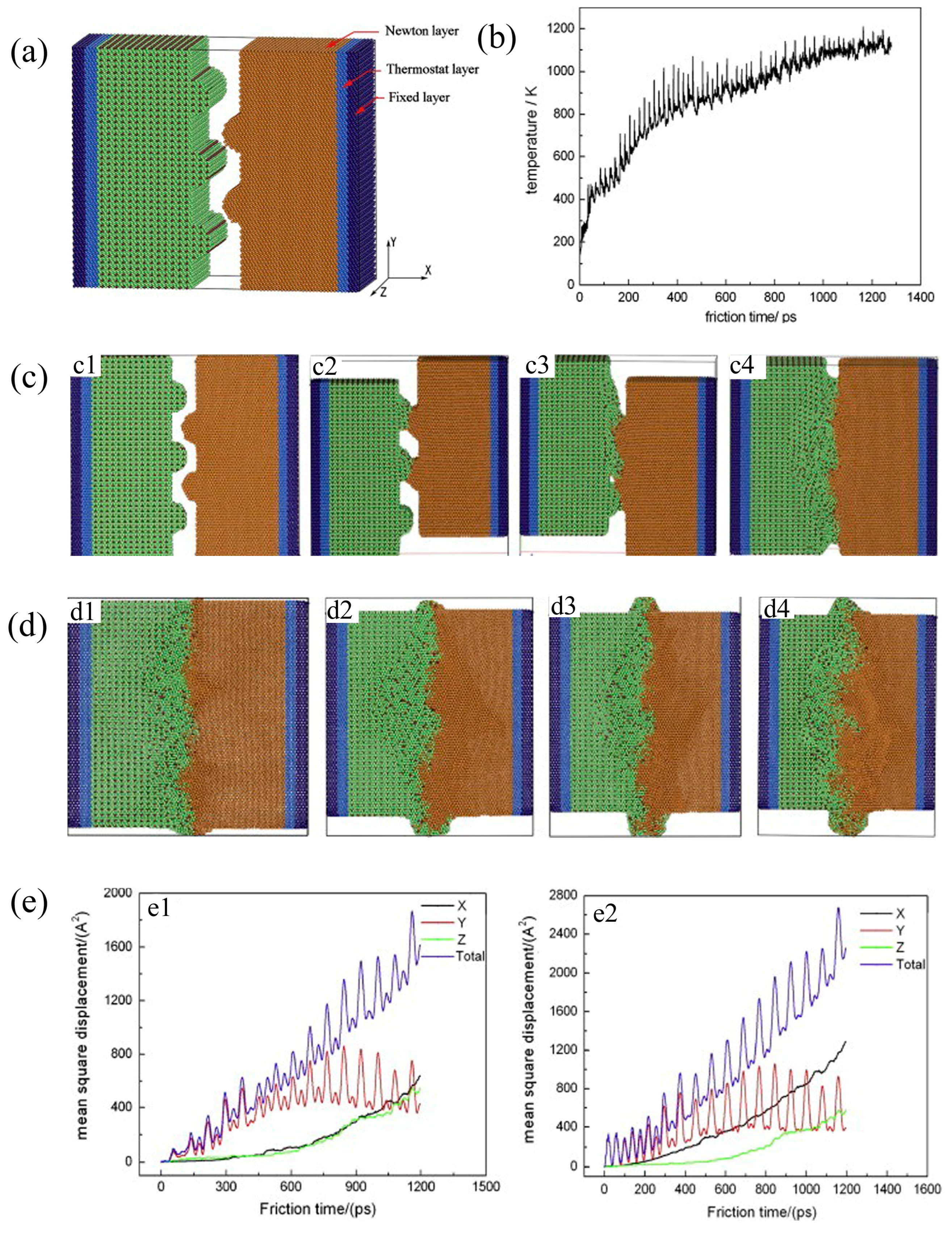
2.4. Simulation of Ti-Al Alloy Welding
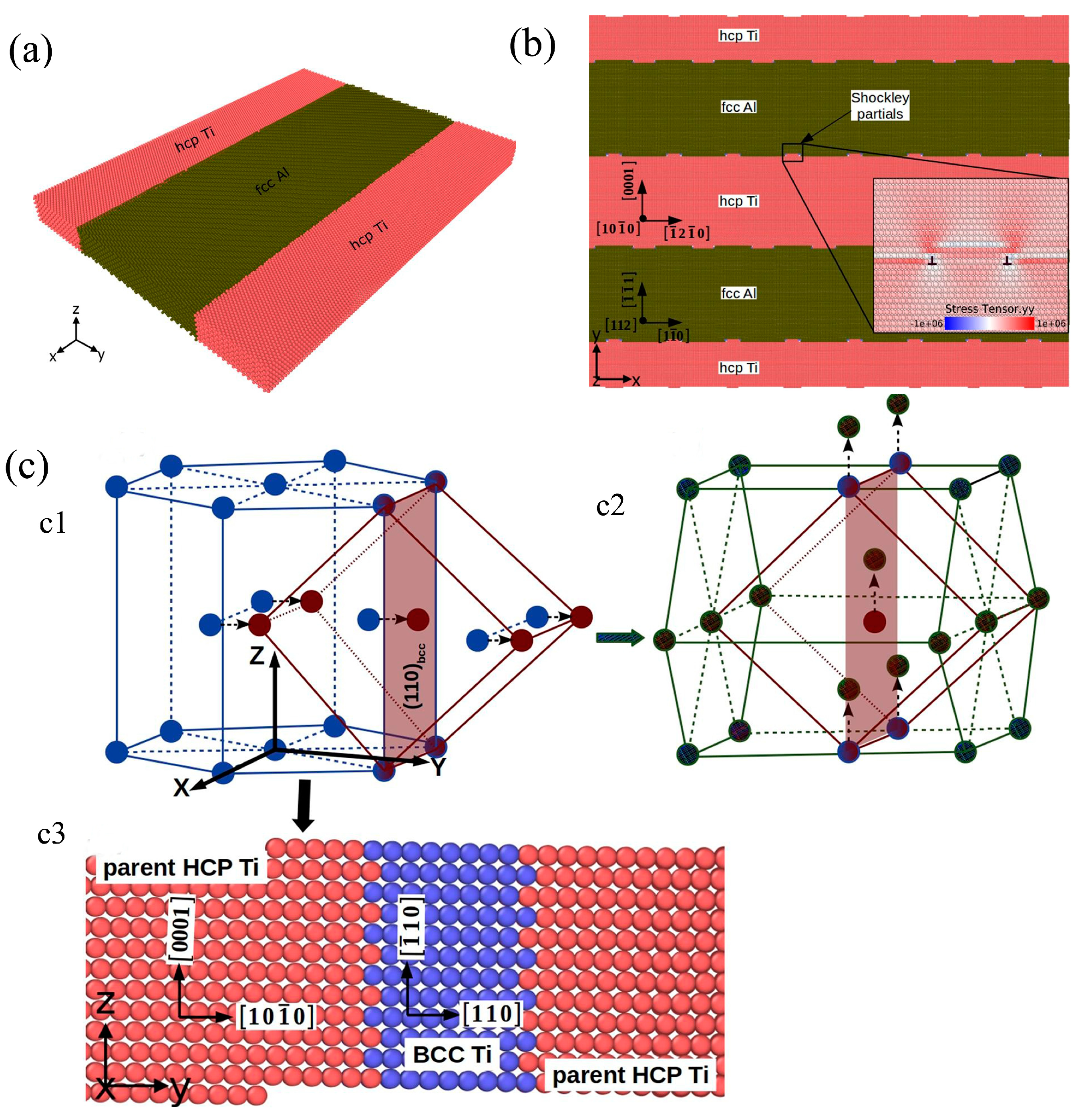
2.5. Simulation of Ti-Al-Based Multi-Layer/Film Materials
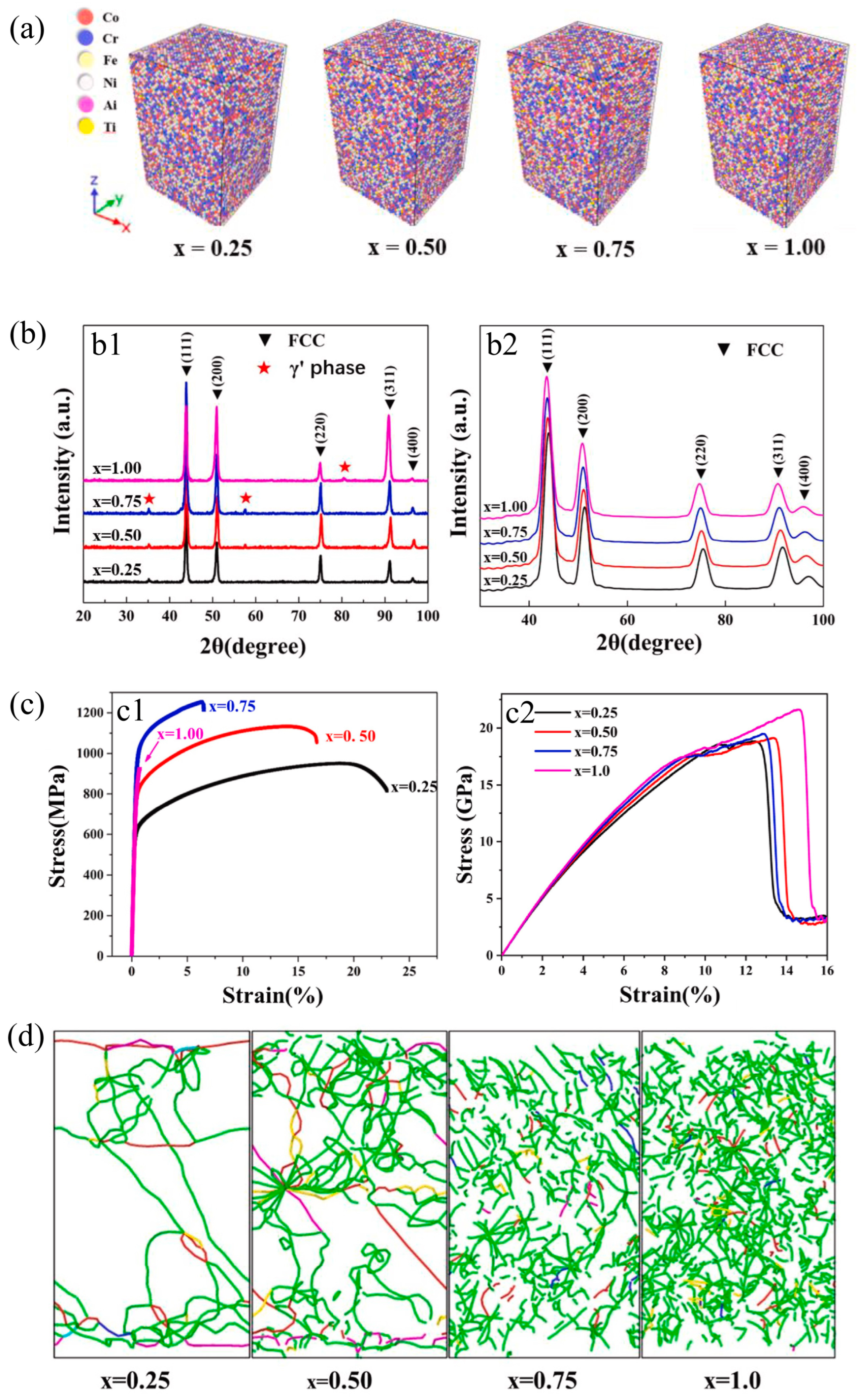
2.6. Others
3. Challenges and Further Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pradeep, G.V.K.; Duraiselvam, M.; Sivaprasad, K. Tribological behavior of laser surface melted γ-TiAl fabricated by electron beam additive manufacturing. J. Mater. Eng. Perform. 2022, 31, 1009–1020. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, Y.; You, X.; Liu, H.; Li, P. Effect of solution heat treatment on the microstructure and hardness of theTi-48Al-2Cr-2Nb alloy prepared by electron beam smelting. J. Mater. Eng. Perform. 2022, 31, 1387–1396. [Google Scholar] [CrossRef]
- Mohammadnejad, A.; Bahrami, A.; Tafaghodi Khajavi, L. Microstructure and mechanical properties of spark plasma sintered nanocrystalline TiAl-x B composites (0.0 < x < 1.5 at.%) containing carbon nanotubes. J. Mater. Eng. Perform. 2021, 30, 4380–4392. [Google Scholar]
- Zha, M.; Wang, H.Y.; Li, S.T.; Li, S.L.; Guan, Q.L.; Jiang, Q.C. Influence of Al addition on the products of self-propagating high-temperature synthesis of Al-Ti-Si system. Mater. Chem. Phys. 2009, 114, 709–715. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, R.; Kou, H.C.; Wang, J.; Zhang, T.B.; Li, J.S.; Zhang, J. Solidification characteristics of high Nb-containing γ-TiAl-based alloys with different aluminum contents. Rare Met. 2015, 34, 381–386. [Google Scholar] [CrossRef]
- Bizot, Q.; Politano, O.; Nepapushev, A.A.; Vadchenko, S.G.; Rogachev, A.S.; Baras, F. Reactivity of the Ti-Al system: Experimental study and molecular dynamics simulations. J. Appl. Phys. 2020, 127, 145304. [Google Scholar] [CrossRef]
- Tian, Y.; Shen, J.; Hu, S.; Gou, J.; Cui, Y. Effects of cold metal transfer mode on the reaction layer of wire and arc additive-manufactured Ti-6Al-4V/Al-6.25 Cu dissimilar alloys. J. Mater. Sci. Technol. 2021, 74, 35–45. [Google Scholar] [CrossRef]
- Rittinghaus, S.K.; Wilms, M.B. Oxide dispersion strengthening of γ-TiAl by laser additive manufacturing. J. Alloys Compd. 2019, 804, 457–460. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, S.; Zhang, Q.K.; Xu, C.; Li, W.D.; Zheng, B.Z.; Hu, F.Q.; Yin, J.; Song, Z.L. In-situ synthesis of Ti-Al intermetallic compounds coating on Ti alloy by magnetron sputtering deposition followed by vacuum annealing. Vacuum 2020, 172, 109060. [Google Scholar] [CrossRef]
- Chen, G.; Peng, Y.; Zheng, G.; Qi, Z.; Wang, M.; Yu, H.; Dong, C.; Liu, C.T. Polysynthetic twinned TiAl single crystals for high-temperature applications. Nat. Mater. 2016, 15, 876–881. [Google Scholar] [CrossRef]
- Shagiev, M.R.; Galeyev, R.M.; Valiakhmetov, O.R.; Safiullin, R.V. Improved mechanical properties of Ti2AlNb-based intermetallic alloys and composites. Adv. Mater. Res. 2009, 59, 105–108. [Google Scholar] [CrossRef]
- Wang, D.P.; Qi, Z.X.; Zhang, H.T.; Chen, G.; Lu, Y.; Sun, B.A.; Liu, C.T. Microscale mechanical properties of ultra-high-strength polysynthetic TiAl-Ti3Al single crystals. Mater. Sci. Eng. A 2018, 732, 14–20. [Google Scholar] [CrossRef]
- Djanarthany, S.; Viala, J.-C.; Bouix, J. An overview of monolithic titanium aluminides based on Ti3Al and TiAl. Mater. Chem. Phys. 2001, 72, 301–319. [Google Scholar] [CrossRef]
- Cao, S.Z.; Xiao, S.L.; Chen, Y.Y.; Xu, L.J.; Wang, X.P.; Han, J.C.; Jia, Y. Phase transformations of the L12-Ti3Al phase in γ-TiAl alloy. Mater. Design. 2017, 121, 61–68. [Google Scholar] [CrossRef]
- Avdeeva, V.; Bazhina, A.; Antipov, M.; Stolin, A.; Bazhin, P. Relationship between structure and properties of intermetallic materials based on γ-TiAl hardened in situ with Ti3Al. Metals 2023, 13, 1002. [Google Scholar] [CrossRef]
- Wimler, D.; Lindemann, J.; Kremmer, T.; Clemens, H.; Mayer, S. Microstructure and mechanical properties of novel TiAl alloys tailored via phase and precipitate morphology. Intermetallics 2021, 138, 107316. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, N.; Liang, H.; Liu, Y. Phase transformation and microstructure control of Ti2AlNb-based alloys: A review. J. Mater. Sci. Technol. 2021, 80, 203–216. [Google Scholar] [CrossRef]
- Hussain, A.; Mehmood, S.; Rasool, N.; Li, N.; Dharmawardhana, C.C. Electronic structure, mechanical and optical properties of TiAl3 (L12 & D022) via first-principles calculations. Chin. J. Phys. 2016, 54, 319–328. [Google Scholar]
- Xie, Y.Q.; Liu, X.B.; Peng, K.; Peng, H.J. Atomic states, potential energies, volumes, stability, and brittleness of ordered FCC TiAl3-type alloys. Phys. B 2004, 353, 15–33. [Google Scholar] [CrossRef]
- Xie, Z.C.; Gao, T.H.; Guo, X.T.; Xie, Q. Molecular dynamics simulation of nanocrystal formation and deformation behavior of Ti3Al alloy. Comp. Mater. Sci. 2015, 98, 245–251. [Google Scholar] [CrossRef]
- Xu, S.; Wan, Q.; Sha, Z.; Liu, Z. Molecular dynamics simulations of nano-indentation and wear of the γTi-Al alloy. Comp. Mater. Sci. 2015, 110, 247–253. [Google Scholar] [CrossRef]
- Poletaev, G.M. Self-diffusion in liquid and solid alloys of the Ti-Al system: Molecular dynamics simulation. J. Exp. Theor. Phys. 2021, 133, 455–460. [Google Scholar] [CrossRef]
- Becquart, C.S.; Decker, K.M.; Domain, C.; Ruste, J.; Souffez, Y.; Turbatte, J.C.; Van Duysen, J.C. Massively parallel molecular dynamics simulations with EAM potentials. Radiat. Eff. Defects Solids 1997, 142, 9–21. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Tuckerman, M.E.; Martyna, G.J. Understanding modern molecular dynamics: Techniques and applications. J. Phys. Chem. B 2000, 104, 159–178. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Zepeda-Ruiz, L.A.; Stukowski, A.; Oppelstrup, T.; Bulatov, V.V. Probing the limits of metal plasticity with molecular dynamics simulations. Nature 2017, 550, 492–495. [Google Scholar] [CrossRef]
- Sankar, N.; Mathew, N.; Sobhan, C.B. Molecular dynamics modeling of thermal conductivity enhancement in metal nanoparticle suspensions. Int. Commun. Heat Mass Transf. 2008, 35, 867–872. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, S.; Sun, D.; Zhang, Y.; Yan, B. Molecular dynamics simulation of mechanical behavior and phase transformation of nanocrystalline NiTi shape memory alloy with gradient structure. Comp. Mater. Sci. 2022, 204, 111186. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, D. Effect of temperature on elastic properties of CNT-polyethylene nanocomposite and its interface using MD simulations. J. Mol. Model. 2018, 24, 178. [Google Scholar] [CrossRef]
- Sri Harish, M.; Patra, P.K. Temperature and its control in molecular dynamics simulations. Mol. Simulat. 2021, 47, 701–729. [Google Scholar] [CrossRef]
- Lu, X.; Ran, H.; Cheng, Q.; Guo, F.; Huang, C. Underlying mechanisms of enhanced plasticity in Ti/Al laminates at elevated temperatures: A molecular dynamics study. J. Mater. Res. Technol. 2024, 28, 31–42. [Google Scholar] [CrossRef]
- Pogorelko, V.V.; Mayer, A.E. Dynamic tensile fracture of iron: Molecular dynamics simulations and micromechanical model based on dislocation plasticity. Int. J. Plast. 2023, 167, 103678. [Google Scholar] [CrossRef]
- Shuang, F.; Aifantis, K.E. Modelling dislocation-graphene interactions in a BCC Fe matrix by molecular dynamics simulations and gradient plasticity theory. Appl. Surf. Sci. 2021, 535, 147602. [Google Scholar] [CrossRef]
- Zhou, J.; Shen, J.; Yue, W.; Liu, Y.; Chen, Z. Molecular dynamics simulation of reinforcement mechanism of graphene/aluminum composites and microstructure evolution. J. Mater. Res. Technol. 2023, 23, 2147–2159. [Google Scholar] [CrossRef]
- Medyanik, S.N.; Shao, S. Strengthening effects of coherent interfaces in nanoscale metallic bilayers. Comp. Mater. Sci. 2009, 45, 1129–1133. [Google Scholar] [CrossRef]
- Shimono, M.; Onodera, H. Molecular dynamics study on formation and crystallization of Ti–Al amorphous alloys. Mater. Sci. Eng. A 2001, 304, 515–519. [Google Scholar] [CrossRef]
- Song, H.; Gao, T.; Gao, Y.; Liu, Y.; Xie, Q.; Chen, Q.; Xiao, Q.Q.; Liang, Y.C.; Wang, B. Hall-Petch relationship in Ti3Al nano-polycrystalline alloys by molecular dynamics simulation. J. Mater. Sci. 2022, 57, 20589–20600. [Google Scholar] [CrossRef]
- Pei, Q.X.; Lu, C.; Fu, M.W. The rapid solidification of Ti3Al: A molecular dynamics study. J. Phys: Condens. Matter 2004, 16, 4203. [Google Scholar] [CrossRef]
- Han, X.J.; Chen, M.; Guo, Z.Y. A molecular dynamics study for the thermophysical properties of liquid Ti–Al alloys. Int. J. Thermophys. 2005, 26, 869–880. [Google Scholar] [CrossRef]
- Polyakova, P.V.; Shcherbinin, S.A.; Baimova, J.A. Molecular dynamics investigation of atomic mixing and mechanical properties of Al/Ti interface. Lett. Mater. 2021, 11, 561–565. [Google Scholar] [CrossRef]
- Gao, T.; He, H.; Liu, Y.; Bian, Z.; Chen, Q.; Xie, Q.; Liang, Y.; Xiao, Q. Molecular dynamics simulation of dislocation network formation and tensile properties of graphene/TiAl-layered composites. Surf. Interfaces 2023, 39, 102983. [Google Scholar] [CrossRef]
- Wu, X.M.; Shi, C.G.; Wang, H.T.; Luo, X.C.; Sun, Z.R. Atomic motion behavior calculation and bonding mechanism analysis of explosive welding of high-strength and high-hardness titanium alloy Ti6Al4V/aluminum alloy 7075. Mater. Res. Express 2023, 10, 106518. [Google Scholar]
- Cui, Z.; Zhou, X.; Meng, Q. Atomic-scale mechanism investigation of mass transfer in laser fabrication process of Ti-Al alloy via molecular dynamics simulation. Metals 2020, 10, 1660. [Google Scholar] [CrossRef]
- Kiselev, S.P.; Zhirov, E.V. Molecular-dynamics simulation of the synthesis of intermetallic Ti–Al. Intermetallics 2014, 49, 106–114. [Google Scholar] [CrossRef]
- Poletaev, G.M.; Bebikhov, Y.V.; Semenov, A.S.; Starostenkov, M.D. Self-diffusion in melts of Ni-Al and Ti-Al systems: Molecular dynamics study. Lett. Mater. 2021, 11, 438–441. [Google Scholar] [CrossRef]
- Guo, F.; Holec, D.; Wang, J.; Li, S.; Du, Y. Impact of V, Hf and Si on oxidation processes in Ti-Al-N: Insights from ab initio molecular dynamics. Surf. Coat. Technol. 2020, 381, 125125. [Google Scholar] [CrossRef]
- Xia, J.H.; Liu, C.S.; Cheng, Z.F.; Shi, D.P. Molecular dynamics simulations on local structure and diffusion in liquid TixAl1−x alloys. Phys. B 2011, 406, 3938–3941. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, X.; Li, C.; Zhou, K. Molecular dynamics simulation on phase transformation of Ti-Al alloy with low Al content. Rare Metal Mater. Eng. 2012, 41, 1010–1015. [Google Scholar] [CrossRef]
- Venkataraman, A.; Shade, P.A.; Adebisi, R.; Sathish, S.; Pilchak, A.L.; Viswanathan, G.B.; Brandes, M.C.; Mills, M.J.; Sangid, M.D. Study of structure and deformation pathways in Ti-7Al using atomistic simulations, experiments, and characterization. Metall. Mater. Trans. A 2017, 48, 2222–2236. [Google Scholar] [CrossRef]
- Arifin, R.; Setiawan, D.R.P.; Triawan, D.; Putra, A.F.S.; Munaji; Winardi, Y.; Putra, W.T.; Darminto. Structural transformation of Ti-based alloys during tensile and compressive loading: An insight from molecular dynamics simulations. MRS Commun. 2023, 13, 225–232. [Google Scholar] [CrossRef]
- Li, T.; Tian, C.; Moridi, A.; Yeo, J. Elucidating interfacial dynamics of Ti-Al systems using molecular dynamics simulation and markov state, odeling. ACS Appl. Mater. Interfaces 2023, 15, 50489–50498. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, L. Investigation on the diffusion behaviors and mechanical properties of the Ti/Al interface using molecular dynamics simulation. J. Mater. Eng. Perform. 2024, 33, 2920–2939. [Google Scholar] [CrossRef]
- Hui, C.; Zhiyuan, R.; Wenke, C.; Ruicheng, F.; Changfeng, Y. Deformation mechanisms in nanotwinned γ-TiAl by molecular dynamics simulation. Mol. Simulat. 2018, 44, 1489–1500. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, L. Molecular dynamics simulation of the tensile deformation behavior of the γ (TiAl)/α2(Ti3Al) interface at different temperatures. J. Mater. Eng. Perform. 2022, 31, 918–932. [Google Scholar] [CrossRef]
- Xu, D.S.; Wang, H.; Yang, R.; Veyssiere, P. Molecular dynamics investigation of deformation twinning in γ-TiAl sheared along the pseudo-twinning direction. Acta Mater. 2008, 56, 1065–1074. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.H. Molecular dynamics simulation studies of benzene, toluene, and p-xylene in NpT ensemble: Thermodynamic, structural, and dynamic properties. Bull.-Korean Chem. Soc. 2002, 23, 447–458. [Google Scholar]
- Wu, H.N.; Xu, D.S.; Wang, H.; Yang, R. Molecular dynamics simulation of tensile deformation and fracture of γ-TiAl with and without surface defects. J. Mater. Sci. Technol. 2016, 32, 1033–1042. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, T.; Gao, Y.; Li, L.; Tan, M.; Xie, Q.; Chen, Q.; Tian, Z.; Liang, Y.; Wang, B. Evolution of defects and deformation mechanisms in different tensile directions of solidified lamellar Ti–Al alloy. Chin. Phys. B 2022, 31, 046105. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, B.; Ou, X.; Ni, S.; Yan, H.; Song, M. Atomic-level study of {10 1} deformation twinning in pure Ti and Ti-5 at.% Al alloy. Int. J. Plast. 2022, 153, 103273. [Google Scholar] [CrossRef]
- Xu, T.T.; Li, J.Y.; Xiao, R.L.; Qin, J.Y.; Ruan, Y.; Li, H. The mixing enthalpy and liquid structural properties of Ti–Al alloys by ab inito molecular dynamics simulation. J. Phase Equilib. Diff. 2022, 43, 585–593. [Google Scholar] [CrossRef]
- Sun, R.; Mi, G.; Huang, X.; Sui, N. Molecular dynamic simulations of Ti-6Al and Fe-12Cr alloys for their heat transfer and oxygen transport behaviors. Mod. Phys. Lett. B 2024, 38, 2350263. [Google Scholar] [CrossRef]
- Feng, R.; Song, W.; Li, H.; Qi, Y.; Qiao, H.; Li, L. Effects of annealing on the residual stress in γ-TiAl alloy by molecular dynamics simulation. Materials 2018, 11, 1025. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, H.; Scheider, I.; Cyron, C.J. Quantifying the high-temperature separation behavior of Lamellar interfaces in γ-titanium aluminide under tensile loading by molecular dynamics. Front. Mater. 2021, 7, 602567. [Google Scholar] [CrossRef]
- Voskoboinikov, R.E. Molecular dynamics simulations of radiation damage in D019 Ti3Al intermetallic compound. Nucl. Instrum. Meth. B 2013, 307, 25–28. [Google Scholar] [CrossRef]
- Shimono, M.; Onodera, H. Molecular dynamics study on liquid-to-amorphous transition in Ti–Al alloys. Mater. Trans. JIM 1998, 39, 147–153. [Google Scholar] [CrossRef]
- Chu, J.J.; Steeves, C.A. Thermal expansion and recrystallization of amorphous Al and Ti: A molecular dynamics study. J. Non-Cryst. Solids 2011, 357, 3765–3773. [Google Scholar] [CrossRef]
- Levchenko, E.V.; Evteev, A.V.; Löwisch, G.G.; Belova, I.V.; Murch, G.E. Molecular dynamics simulation of alloying in a Ti-coated Al nanoparticle. Intermetallics 2012, 22, 193–202. [Google Scholar] [CrossRef]
- Levchenko, E.V.; Evteev, A.V.; Lorscheider, T.; Belova, I.V.; Murch, G.E. Molecular dynamics simulation of alloying in an Al-coated Ti nanoparticle. Comp. Mater. Sci. 2013, 79, 316–325. [Google Scholar] [CrossRef]
- Tahiri, M.; Hassani, A.; Sbiaai, K.; Hasnaoui, A. Investigating local atomic structural order in TiAl3 metallic glass using molecular dynamic simulation. Comput. Condens. Matter 2018, 14, 74–83. [Google Scholar] [CrossRef]
- Xie, Z.C.; Gao, T.H.; Guo, X.T.; Qin, X.M.; Xie, Q. Glass formation and icosahedral medium-range order in liquid Ti-Al alloys. Comp. Mater. Sci. 2014, 95, 502–508. [Google Scholar] [CrossRef]
- Han, X.; Liu, P.; Sun, D.; Wang, Q. Quantifying the role of interface atomic structure in the compressive response of Ti2AlN/TiAl composite using MD simulations. J. Mater. Sci. 2019, 54, 5536–5550. [Google Scholar] [CrossRef]
- Duong, T.C.; Singh, N.; Arróyave, R. First-principles calculations of finite-temperature elastic properties of Ti2AlX (X=C or N). Comp. Mater. Sci. 2013, 79, 296–302. [Google Scholar] [CrossRef]
- Han, X.; Liu, P.; Sun, D.; Wang, Q. Molecular dynamics simulations of the tensile responses and fracture mechanisms of Ti2AlN/TiAl composite. Theor. Appl. Fract. Mech. 2019, 101, 217–223. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Li, Q.; Gong, M.; Xue, S.; Ding, J.; Li, J.; Cho, J.; Niu, T.; Su, R.; Richter, N.A. Deformation behavior and phase transformation of nanotwinned Al/Ti multilayers. Appl. Surf. Sci. 2020, 527, 146776. [Google Scholar] [CrossRef]
- Li, P.; Wang, L.; Yan, S.; Meng, M.; Xue, K. Temperature effect on the diffusion welding process and mechanism of B2-O interface in the Ti2AlNb-based alloy: A molecular dynamics simulation. Vacuum 2020, 173, 109118. [Google Scholar] [CrossRef]
- Ou, P.; Cao, Z.; Rong, J.; Yu, X. Molecular dynamics study on the welding behavior in dissimilar TC4-TA17 titanium alloys. Materials 2022, 15, 5606. [Google Scholar] [CrossRef]
- Zhang, H.; Su, Y.C.; Han, Y.; Jiang, S. Molecular dynamics study of melting behavior of planar stacked Ti–Al core–shell nanoparticles. J. Compos. Sci. 2022, 6, 126. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Xia, X.; Zhang, Z.; Wang, T.; Zhang, H. A molecular dynamics simulation-based laser melting behavior analysis for Ti–Al binary alloy. Int. J. Mod. Phys. B 2023, 37, 2350196. [Google Scholar] [CrossRef]
- Moon, S.; Paek, J.H.; Jang, Y.H.; Kang, K. Mixing behavior of Ti–Al interface during the ultrasonic welding process and its welding strength: Molecular dynamics study. Heliyon 2024, 10, e25116. [Google Scholar] [CrossRef]
- Song, C.; Lin, T.; He, P.; Jiao, Z.; Tao, J.; Ji, Y. Molecular dynamics simulation of linear friction welding between dissimilar Ti-based alloys. Comp. Mater. Sci. 2014, 83, 35–38. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, C. Strengthening and toughening mechanisms of amorphous/amorphous nanolaminates. Int. J. Plast. 2016, 80, 75–85. [Google Scholar] [CrossRef]
- Salehinia, I.; Wang, J.; Bahr, D.F.; Zbib, H.M. Molecular dynamics simulations of plastic deformation in Nb/NbC multilayers. Int. J. Plast. 2014, 59, 119–132. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Su, R.; Niu, T.J.; Richter, N.A.; Xue, S.; Li, Q.; Ding, J.; Yang, B.; Wang, H.; Zhang, X. Thermal stability and deformability of annealed nanotwinned Al/Ti multilayers. Scr. Mater. 2020, 186, 219–224. [Google Scholar] [CrossRef]
- Wang, C.; Shi, K.; Gross, C.; Pureza, J.M.; de Mesquita Lacerda, M.; Chung, Y.W. Toughness enhancement of nanostructured hard coatings: Design strategies and toughness measurement techniques. Surf. Coat. Technol. 2014, 257, 206–212. [Google Scholar] [CrossRef]
- Shugurov, A.R.; Panin, A.V.; Dmitriev, A.I.; Nikonov, A.Y. Multiscale fracture of Ti-Al-N coatings under uniaxial tension. Phys. Mesomech. 2021, 24, 185–195. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Kong, Y.; Chen, L.; Du, Y. Influence of oxygen addition on the oxidation resistance of TiAlN. Scr. Mater. 2023, 224, 115148. [Google Scholar] [CrossRef]
- Tang, G.; Ou, Z.; Liu, F.; Li, T.; Su, F.; Zheng, J.; Liang, Z. Enhancing wear resistance of aluminum alloy by fabricating a Ti-Al modified layer via surface mechanical attrition treatment. Tribol. Int. 2024, 193, 109462. [Google Scholar] [CrossRef]
- Maurya, S.K.; Nie, J.F.; Alankar, A. Atomistic analyses of HCP-FCC transformation and reorientation of Ti in Al-Ti multilayers. Comp. Mater. Sci. 2021, 192, 110329. [Google Scholar] [CrossRef]
- Chen, P.; Wang, F.; Li, B. Transitory phase transformations during {102} twinning in titanium. Acta Mater. 2019, 171, 65–78. [Google Scholar] [CrossRef]
- George, E.P.; Raabe, D.; Ritchie, R.O. High-entropy alloys. Nat. Rev. Mater. 2019, 4, 515–534. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Li, W.; Xie, D.; Li, D.; Zhang, Y.; Gao, Y.; Liaw, P.K. Mechanical behavior of high-entropy alloys. Prog. Mater. Sci. 2021, 118, 100777. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, Y.L.; Lin, S.J.; Chen, S.K. High-entropy alloys—A new era of exploitation. Mater. Sci. Forum 2007, 560, 1–9. [Google Scholar] [CrossRef]
- Zhang, W.; Liaw, P.K.; Zhang, Y. Science and technology in high-entropy alloys. Sci. China Mater. 2018, 61, 2–22. [Google Scholar] [CrossRef]
- Li, J.; Fang, Q.; Liu, B.; Liu, Y.; Liu, Y. Mechanical behaviors of AlCrFeCuNi high-entropy alloys under uniaxial tension via molecular dynamics simulation. RSC Adv. 2016, 6, 76409–76419. [Google Scholar] [CrossRef]
- Chen, K.T.; Wei, T.J.; Li, G.C.; Chen, M.Y.; Chen, Y.S.; Chang, S.W.; Yen, H.W.; Chen, C.S. Mechanical properties and deformation mechanisms in CoCrFeMnNi high entropy alloys: A molecular dynamics study. Mater. Chem. Phys. 2021, 271, 124912. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, P.; Qiu, J.; Sun, W.; Saikov, I.; Shcherbakov, V.; Alymov, M. Microstructural evolution and mechanical properties of AlxCoCrFeNi high-entropy alloys under uniaxial tension: A molecular dynamics simulations study. Mater. Today Commun. 2021, 28, 102525. [Google Scholar] [CrossRef]
- Sun, Z.H.; Zhang, J.; Xin, G.X.; Xie, L.; Yang, L.C.; Peng, Q. Tensile mechanical properties of CoCrFeNiTiAl high entropy alloy via molecular dynamics simulations. Intermetallics 2022, 142, 107444. [Google Scholar] [CrossRef]
- Zheng, G.; Chen, Y.; Xiang, H.; Zhang, J.; Tu, K.N.; Feng, C.; Li, P.; Song, L.; Sha, G.; Qi, Z. Coupled nucleation of dual-phase lamellar structure. Innov. Mater. 2023, 1, 100043. [Google Scholar] [CrossRef]
- Li, Z.; Feng, Y.; Wen, Y.; Peng, X.; Cai, Z.; Peng, C.; Wang, R. Ab initio molecular dynamics study on the local structures and solid/liquid interface in liquid Al-Ti and Al-B-Ti alloys. Mater. Today Commun. 2024, 39, 109290. [Google Scholar] [CrossRef]
- Elkhateeb, M.G.; Shin, Y.C. Molecular dynamics-based cohesive zone representation of Ti6Al4V/TiC composite interface. Mater. Des. 2018, 155, 161–169. [Google Scholar] [CrossRef]
- Pei, Q.X.; Jhon, M.H.; Quek, S.S.; Wu, Z. A systematic study of interatomic potentials for mechanical behaviours of Ti-Al alloys. Comp. Mater. Sci. 2021, 188, 110239. [Google Scholar] [CrossRef]
- Zhai, B.; Wang, H.P. Accurate interatomic potential for the nucleation in liquid Ti-Al binary alloy developed by deep neural network learning method. Comp. Mater. Sci. 2023, 216, 111843. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance | Ti-Based Alloys | TiAl-Based Alloys | Ti3Al-Based Alloys |
|---|---|---|---|
| Crystal structure | hcp/bcc | L10 | D019 |
| Density/g·cm−3 | 4.5 | 4.1–4.7 | 3.7~4.3 |
| Elastic modulus/GPa | 96–115 | 100~145 | 160~180 |
| Yield strength/MPa | 380–1150 | 700~990 | 400~800 |
| Tensile strength/MPa | 480~1200 | 800~1140 | 450~1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Hao, Z.; Xu, L.; Tang, M.; Wei, L.; Wang, L. A Review of Molecular Dynamics Simulation of Different Ti-Al-Based Alloys. Metals 2024, 14, 1018. https://doi.org/10.3390/met14091018
Li N, Hao Z, Xu L, Tang M, Wei L, Wang L. A Review of Molecular Dynamics Simulation of Different Ti-Al-Based Alloys. Metals. 2024; 14(9):1018. https://doi.org/10.3390/met14091018
Chicago/Turabian StyleLi, Ningning, Zhenjie Hao, Lei Xu, Mingqi Tang, Leyu Wei, and Lifei Wang. 2024. "A Review of Molecular Dynamics Simulation of Different Ti-Al-Based Alloys" Metals 14, no. 9: 1018. https://doi.org/10.3390/met14091018
APA StyleLi, N., Hao, Z., Xu, L., Tang, M., Wei, L., & Wang, L. (2024). A Review of Molecular Dynamics Simulation of Different Ti-Al-Based Alloys. Metals, 14(9), 1018. https://doi.org/10.3390/met14091018