Whole-Genome Sequencing Analysis of Salmonella Enterica Serotype Enteritidis Isolated from Poultry Sources in South Korea, 2010–2017

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Detection of Antimicrobial Resistance Genes and Mutations in the gyrA, gyrB, parC and parE Genes
2.1.1. Aminoglycoside Resistance Genes
2.1.2. Sulfonamide and Trimethoprim Resistance Genes
2.1.3. Beta-Lactam Resistance Genes
2.1.4. Tetracycline Resistance Genes
2.1.5. Phenicol Resistance Genes
2.1.6. Detection of Mutations in the gyrA, gyrB, parC and parE Genes
2.2. Virulence Gene Profiles
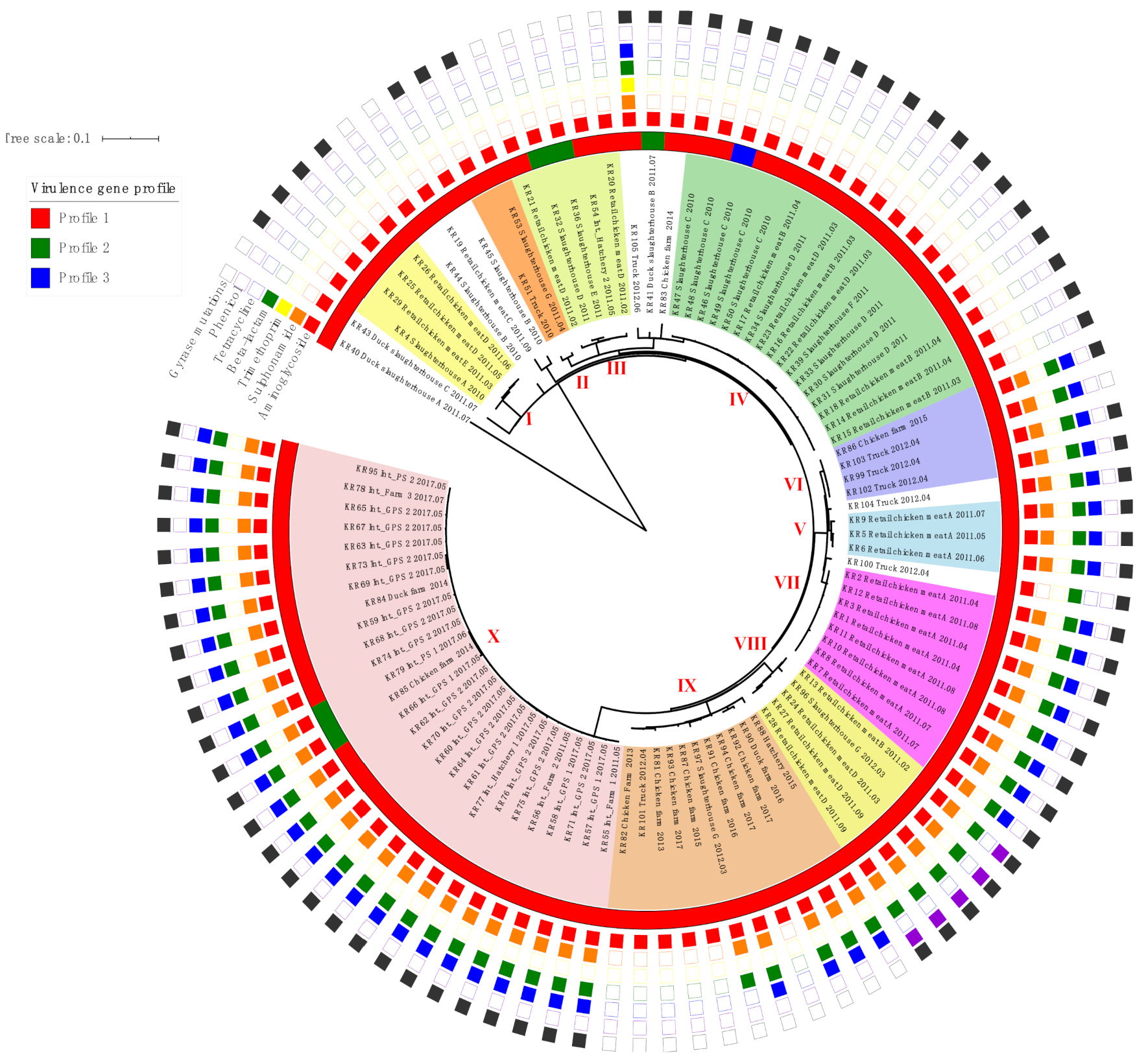
2.3. Phylogenetic Analysis of SE Isolates
3. Discussion
4. Materials and Methods
4.1. Bacterial Isolates
4.2. Genome Sequencing
4.3. Sequencing Reads Trimming, Filtering, and Classification
4.4. De Novo Assembly and Serotyping
4.5. Antibiotic Resistance Analysis
4.6. Virulence Genes Investigation
4.7. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States--major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar]
- Leekitcharoenphon, P.; Hendriksen, R.S.; Le Hello, S.; Weill, F.X.; Baggesen, D.L.; Jun, S.R.; Ussery, D.W.; Lund, O.; Crook, D.W.; Wilson, D.J.; et al. Global Genomic Epidemiology of Salmonella enterica Serovar Typhimurium DT104. Appl. Environ. Microbiol. 2016, 82, 2516–2526. [Google Scholar]
- Ferrari, R.G.; Rosario, D.K.A.; Cunha-Neto, A.; Mano, S.B.; Figueiredo, E.E.S.; Conte-Junior, C.A. Worldwide Epidemiology of Salmonella Serovars in Animal-Based Foods: A Meta-analysis. Appl. Environ. Microbiol. 2019, 85, e00591-19. [Google Scholar]
- Choi, S.W.; Ha, J.S.; Kim, B.Y.; Lee, D.H.; Park, J.K.; Youn, H.N.; Hong, Y.H.; Lee, S.B.; Lee, J.B.; Park, S.Y.; et al. Prevalence and characterization of Salmonella species in entire steps of a single integrated broiler supply chain in Korea. Poult. Sci. 2014, 93, 1251–1257. [Google Scholar]
- Deng, X.; Desai, P.T.; den Bakker, H.C.; Mikoleit, M.; Tolar, B.; Trees, E.; Hendriksen, R.S.; Frye, J.G.; Porwollik, S.; Weimer, B.C.; et al. Genomic epidemiology of Salmonella enterica serotype Enteritidis based on population structure of prevalent lineages. Emerg. Infect. Dis. 2014, 20, 1481–1489. [Google Scholar]
- Lee, D.H.; Hyeon, J.Y.; Kim, J.; Kim, J.S.; Kim, S.J.; Jeon, S.E.; Choi, S.W.; Hong, W.T.; Song, C.S.; Lee, S.W. Close genetic relationship between Salmonella enterica serovar Enteritidis isolated from patients with diarrhoea and poultry in the Republic of Korea. Clin. Microbiol. Infect. 2015, 21, e68–e70. [Google Scholar]
- Toro, M.; Retamal, P.; Ayers, S.; Barreto, M.; Allard, M.; Brown, E.W.; Gonzalez-Escalona, N. Whole-Genome Sequencing Analysis of Salmonella enterica Serovar Enteritidis Isolates in Chile Provides Insights into Possible Transmission between Gulls, Poultry, and Humans. Appl. Environ. Microbiol. 2016, 82, 6223–6232. [Google Scholar]
- Lienau, E.K.; Strain, E.; Wang, C.; Zheng, J.; Ottesen, A.R.; Keys, C.E.; Hammack, T.S.; Musser, S.M.; Brown, E.W.; Allard, M.W.; et al. Identification of a salmonellosis outbreak by means of molecular sequencing. N. Engl. J. Med. 2011, 364, 981–982. [Google Scholar]
- Eppinger, M.; Mammel, M.K.; Leclerc, J.E.; Ravel, J.; Cebula, T.A. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. USA 2011, 108, 20142–20147. [Google Scholar]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 2011, 365, 709–717. [Google Scholar]
- Deng, X.; den Bakker, H.C.; Hendriksen, R.S. Genomic Epidemiology: Whole-Genome-Sequencing-Powered Surveillance and Outbreak Investigation of Foodborne Bacterial Pathogens. Annu. Rev. Food Sci. Technol. 2016, 7, 353–374. [Google Scholar]
- Carleton, H.A.; Gerner-Smidt, P. Whole-genome sequencing is taking over foodborne disease surveillance. Microbe Mag. 2016, 11, 311–317. [Google Scholar]
- Tolar, B.; Joseph, L.A.; Schroeder, M.N.; Stroika, S.; Ribot, E.M.; Hise, K.B.; Gerner-Smidt, P. An Overview of PulseNet USA Databases. Foodborne Pathog. Dis. 2019, 16, 457–462. [Google Scholar]
- Almeida, F.; Medeiros, M.I.; Kich, J.D.; Falcao, J.P. Virulence-associated genes, antimicrobial resistance and molecular typing of Salmonella Typhimurium strains isolated from swine from 2000 to 2012 in Brazil. J. Appl. Microbiol. 2016, 120, 1677–1690. [Google Scholar]
- Almeida, F.; Seribelli, A.A.; Medeiros, M.I.C.; Rodrigues, D.D.P.; de MelloVarani, A.; Luo, Y.; Allard, M.W.; Falcao, J.P. Phylogenetic and antimicrobial resistance gene analysis of Salmonella Typhimurium strains isolated in Brazil by whole genome sequencing. PLoS ONE 2018, 13, e0201882. [Google Scholar]
- McDermott, P.F.; Tyson, G.H.; Kabera, C.; Chen, Y.; Li, C.; Folster, J.P.; Ayers, S.L.; Lam, C.; Tate, H.P.; Zhao, S. Whole-Genome Sequencing for Detecting Antimicrobial Resistance in Nontyphoidal Salmonella. Antimicrob. Agents Chemother 2016, 60, 5515–5520. [Google Scholar]
- Pearce, M.E.; Alikhan, N.F.; Dallman, T.J.; Zhou, Z.; Grant, K.; Maiden, M.C.J. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 2018, 274, 1–11. [Google Scholar]
- Pornsukarom, S.; van Vliet, A.H.M.; Thakur, S. Whole genome sequencing analysis of multiple Salmonella serovars provides insights into phylogenetic relatedness, antimicrobial resistance, and virulence markers across humans, food animals and agriculture environmental sources. BMC Genom. 2018, 19, 801. [Google Scholar]
- Simon, S.; Trost, E.; Bender, J.; Fuchs, S.; Malorny, B.; Rabsch, W.; Prager, R.; Tietze, E.; Flieger, A. Evaluation of WGS based approaches for investigating a food-borne outbreak caused by Salmonella enterica serovar Derby in Germany. Food Microbiol. 2018, 71, 46–54. [Google Scholar]
- Tay, M.Y.F.; Pathirage, S.; Chandrasekaran, L.; Wickramasuriya, U.; Sadeepanie, N.; Waidyarathna, K.D.K.; Liyanage, L.D.C.; Seow, K.L.G.; Hendriksen, R.S.; Takeuchi, M.T.; et al. Whole-Genome Sequencing Analysis of Nontyphoidal Salmonella enterica of Chicken Meat and Human Origin Under Surveillance in Sri Lanka. Foodborne Pathog. Dis. 2019, 16, 531–537. [Google Scholar]
- Kim, S.; Kim, E.; Park, S.; Hahn, T.W.; Yoon, H. Genomic Approaches for Understanding the Characteristics of Salmonella enterica subsp. enterica Serovar Typhimurium ST1120, Isolated from Swine Feces in Korea. J. Microbiol. Biotech. 2017, 27, 1983–1993. [Google Scholar]
- Park, A.K.; Shin, E.; Kim, S.; Park, J.; Jeong, H.J.; Chun, J.H.; Hwang, K.J.; Kim, J. The Traveler-associated High-level Ciprofloxacin-resistant Salmonella enterica Serovar Kentucky in the Republic of Korea. J. Global Antimicrobial. Resist. 2019. [Google Scholar] [CrossRef]
- Woojung, L.; Sewook, P.; Ran Hee, Y.; In-Sun, J.; Hyo Sun, K.; Soon Han, K. Complete genome sequence of Salmonella Enteritidis MFDS1004839 isolated from food. Korean J. Microbiol. 2018, 54, 164–166. [Google Scholar]
- Brown, E.; Dessai, U.; McGarry, S.; Gerner-Smidt, P. Use of Whole-Genome Sequencing for Food Safety and Public Health in the United States. Foodborne Pathog. Dis. 2019, 16, 441–450. [Google Scholar]
- Pightling, A.W.; Pettengill, J.B.; Luo, Y.; Baugher, J.D.; Rand, H.; Strain, E. Interpreting Whole-Genome Sequence Analyses of Foodborne Bacteria for Regulatory Applications and Outbreak Investigations. Front. Microbiol. 2018, 9, 1482. [Google Scholar]
- Kim, M.S.; Lim, T.H.; Jang, J.H.; Lee, D.H.; Kim, B.Y.; Kwon, J.H.; Choi, S.W.; Noh, J.Y.; Hong, Y.H.; Lee, S.B.; et al. Prevalence and antimicrobial resistance of Salmonella species isolated from chicken meats produced by different integrated broiler operations in Korea. Poult Sci. 2012, 91, 2370–2375. [Google Scholar]
- Lee, S.K.; Choi, D.; Kim, H.S.; Kim, D.H.; Seo, K.H. Prevalence, Seasonal Occurrence, and Antimicrobial Resistance of Salmonella spp. Isolates Recovered from Chicken Carcasses Sampled at Major Poultry Processing Plants of South Korea. Foodborne Pathog. Dis. 2016, 13, 544–550. [Google Scholar]
- Yoon, R.H.; Cha, S.Y.; Wei, B.; Roh, J.H.; Seo, H.S.; Oh, J.Y.; Jang, H.K. Prevalence of Salmonella isolates and antimicrobial resistance in poultry meat from South Korea. J. Food Prot. 2014, 77, 1579–1582. [Google Scholar]
- Cardoso, M.O.; Ribeiro, A.R.; Santos, L.R.D.; Pilotto, F.; Moraes, H.L.S.d.; Salle, C.T.P.; Rocha, S.L.d.S.; Nascimento, V.P.d. Antibiotic resistance in Salmonella Enteritidis isolated from broiler carcasses. Brazilian J. Microbiol. 2006, 37, 368–371. [Google Scholar]
- Thung, T.Y.; Mahyudin, N.A.; Basri, D.F.; Wan Mohamed Radzi, C.W.; Nakaguchi, Y.; Nishibuchi, M.; Radu, S. Prevalence and antibiotic resistance of Salmonella Enteritidis and Salmonella Typhimurium in raw chicken meat at retail markets in Malaysia. Poult Sci. 2016, 95, 1888–1893. [Google Scholar]
- Singh, R.; Yadav, A.S.; Tripathi, V.; Singh, R.P. Antimicrobial resistance profile of Salmonella present in poultry and poultry environment in north India. Food Control. 2013, 33, 545–548. [Google Scholar]
- Zhou, X.; Xu, L.; Xu, X.; Zhu, Y.; Suo, Y.; Shi, C.; Shi, X. Antimicrobial Resistance and Molecular Characterization of Salmonella enterica Serovar Enteritidis from Retail Chicken Products in Shanghai, China. Foodborne Pathog. Dis. 2018, 15, 346–352. [Google Scholar]
- Diarra, M.S.; Delaquis, P.; Rempel, H.; Bach, S.; Harlton, C.; Aslam, M.; Pritchard, J.; Topp, E. Antibiotic resistance and diversity of Salmonella enterica serovars associated with broiler chickens. J. Food Prot. 2014, 77, 40–49. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Halatsi, K.; Oikonomou, I.; Lambiri, M.; Mandilara, G.; Vatopoulos, A.; Kyriacou, A. PCR detection of Salmonella spp. using primers targeting the quorum sensing gene sdiA. FEMS Microbiol. Lett. 2006, 259, 201–207. [Google Scholar]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar]
- Zhang, S.; Yin, Y.; Jones, M.B.; Zhang, Z.; Deatherage Kaiser, B.L.; Dinsmore, B.A.; Fitzgerald, C.; Fields, P.I.; Deng, X. Salmonella serotype determination utilizing high-throughput genome sequencing data. J. Clin. Microbiol. 2015, 53, 1685–1692. [Google Scholar]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother 2012, 67, 2640–2644. [Google Scholar]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic. Acids. Res. 2017, 45, D535–D542. [Google Scholar]
- Patchanee, P.; Eiamsam-Ang, T.; Vanaseang, J.; Boonkhot, P.; Tadee, P. Determination of regional relationships among Salmonella spp. isolated from retail pork circulating in the Chiang Mai municipality area using a WGS data approach. Int. J. Food Microbiol. 2017, 254, 18–24. [Google Scholar]
- Davis, S.P.J.; Luo, Y.; Payne, J.; Shpuntoff, A.; Rand, H.; Strain, E. CFSAN SNP Pipeline: An automated method for constructing SNP matrices from next-generation sequence data. PeerJ. Computer Sci. 2015, 1, e20. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar]
- Zhou, Z.; Alikhan, N.F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carrico, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome. Res. 2018, 28, 1395–1404. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Pattern | Antimicrobial Genes (Identity, %) | Chromosome Mutations | Number of Isolates (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Aminoglycoside | Sulfonamide | Trimethoprim | Beta-Lactam | Tetracycline | Phenicol | |||
| Pattern 1 | aac(6’)-Iaa (96.4), aph(3’’)-Ib (100), aph(6)-Id (99.76-100) | sul2 (100) | blaTEM-1B (99.9-100) | tet(A) (99.9-100) | gyrA p.D87N | 27 (28.1) | ||
| Pattern 2 | aac(6’)-Iaa (96.4), aph(3’’)-Ib (100), aph(6)-Id (99.76-100) | sul2 (100) | blaTEM-1B (99.9-100) | tet(A) (99.9-100) | - | 4 (4.2) | ||
| Pattern 3 | aac(6’)-Iaa (96.4), aph(3’’)-Ib (100), aph(6)-Id (100) | sul2 (100) | blaTEM-1B (100) | catA2 (96.1) | gyrA p.D87G | 5 (5.2) | ||
| Pattern 4 | aac(6’)-Iaa (96.4), aph(3’’)-Ib (100), aph(6)-Id (99.8) | sul2 (100) | blaTEM-1B (100) | - | 1 (1.0) | |||
| Pattern 5 | aac(3)-Iid (99.9), aac(6’)-Iaa (96.4), aph(3’’)-Ib (99.9), aph(3’)-Ia (100), aph(6)-Id (100) | sul2 (100) | blaCTX-M-15 (100) | tet(A) (97.3-100) | gyrA p.D87N | 15 (15.6) | ||
| Pattern 6 | aac(3)-Iid (99.9), aac(6’)-Iaa (96.4), aph(3’’)-Ib (99.9), aph(3’)-Ia (100), aph(6)-Id (100) | sul2 (100) | blaCTX-M-15 (100) | tet(A) (97.3-100) | - | 1 (1.0) | ||
| Pattern 7 | aac(6’)-Iaa (96.4) | sul2 (100) | dfrA1 (95.8) | blaTEM-1B (100) | - | 1 (1.0) | ||
| Pattern 8 | aac(6’)-Iaa (96.4) | Sul1 (100) | dfrA1 (95.4) | blaTEM-1B (100) | tet(A) (98.8) | gyrA p.D87G | 1 (1.0) | |
| Pattern 9 | aac(6’)-Iaa (96.4) | gyrA p.D87N | 7 (7.3) | |||||
| Pattern 10 | aac(6’)-Iaa (96.4) | gyrA p.D87G | 19 (19.8) | |||||
| Pattern 11 | aac(6’)-Iaa (96.4) | - | 14 (14.6) | |||||
| Pattern 12 | aac(3)-Iid (99.9), aac(6’)-Iaa (96.4) | blaCTX-M-15 (99.7) | gyrA p.D87N | 1 (1.0) | ||||
| Number of isolates (%) | 96 (100) | 55 (57.3) | 2 (2.1) | 56 (58.3) | 48 (50%) | 1 (1.0) | 75 (78.1) | |
| Profile | Virulence Genes | Number of Isolates(%) | |||||
|---|---|---|---|---|---|---|---|
| Adhesion Effectors (sef, pef, sfm and fim) | Invasion Effectors (sop, inv., org, sip, spa, sif, fli, flg, hil and spr) | Host Cell Survival Effectors (ssa, sse, prg and pag) | Virulence Plasmid Gene (spv) | Enterotoxin-Producing Gene (stn) | Biofilm Regulator (bss) | ||
| Profile 1 | Pef, fim | sop, inv., org, sip, spa, sif, fli, flg, hil | ssa, sse, prg, pag | spv | - | - | 90 (93.8) |
| Profile 2 | Fim | sop, inv., org, sip, spa, sif, fli, flg, hil | ssa, sse, prg, pag | - | - | - | 5 (5.2) |
| Profile 3 | Pef, fim | sop, inv., org, sip, spa, sif, fli, flg, hil | ssa, sse, prg, pag | - | - | - | 1 (1.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyeon, J.-Y.; Li, S.; Mann, D.A.; Zhang, S.; Kim, K.-J.; Lee, D.-H.; Deng, X.; Song, C.-S. Whole-Genome Sequencing Analysis of Salmonella Enterica Serotype Enteritidis Isolated from Poultry Sources in South Korea, 2010–2017. Pathogens 2021, 10, 45. https://doi.org/10.3390/pathogens10010045
Hyeon J-Y, Li S, Mann DA, Zhang S, Kim K-J, Lee D-H, Deng X, Song C-S. Whole-Genome Sequencing Analysis of Salmonella Enterica Serotype Enteritidis Isolated from Poultry Sources in South Korea, 2010–2017. Pathogens. 2021; 10(1):45. https://doi.org/10.3390/pathogens10010045
Chicago/Turabian StyleHyeon, Ji-Yeon, Shaoting Li, David A. Mann, Shaokang Zhang, Kyu-Jik Kim, Dong-Hun Lee, Xiangyu Deng, and Chang-Seon Song. 2021. "Whole-Genome Sequencing Analysis of Salmonella Enterica Serotype Enteritidis Isolated from Poultry Sources in South Korea, 2010–2017" Pathogens 10, no. 1: 45. https://doi.org/10.3390/pathogens10010045
APA StyleHyeon, J. -Y., Li, S., Mann, D. A., Zhang, S., Kim, K. -J., Lee, D. -H., Deng, X., & Song, C. -S. (2021). Whole-Genome Sequencing Analysis of Salmonella Enterica Serotype Enteritidis Isolated from Poultry Sources in South Korea, 2010–2017. Pathogens, 10(1), 45. https://doi.org/10.3390/pathogens10010045