Abstract
The main pathogenic factor of Bacillus anthracis is a three-component toxin encoded by the pagA, lef, and cya genes, which are located on the pXO1 plasmid. The atxA gene, which encodes the primary regulator of pathogenicity factor expression, is located on the same plasmid. In this work, we evaluated the polymorphism of the pagA, lef, cya, and atxA genes for 85 B. anthracis strains from different evolutionary lineages and canSNP groups. We have found a strong correlation of 19 genotypes with the main evolutionary lineages, but the correlation with the canSNP group of the strain was not as strong. We have detected several genetic markers indicating the geographical origin of the strains, for example, their source from the steppe zone of the former USSR. We also found that strains of the B.Br.001/002 group caused an anthrax epidemic in Russia in 2016 and strains isolated during paleontological excavations in the Russian Arctic have the same genotype as the strains of the B.Br.CNEVA group circulating in Central Europe. This data could testify in favor of the genetic relationship of these two groups of strains and hypothesize the ways of distribution of their ancestral forms between Europe and the Arctic.
1. Introduction
Anthrax is a particularly dangerous infection caused by the spore-forming Gram-positive bacterium Bacillus anthracis [1]. The main feature of the systematic position of B. anthracis is its high genetic similarity with some other bacilli—B. cereus, B. thuringiensis, B. mycoides, B. pseudomycoides, B. weihenstephanensis, B. cytotoxicus, and B. toyonensis. These species form a group called Bacillus cereus sensu lato, or Bacillus cereus complex [2]. Their genetic similarity provides grounds for considering this entire group as one unique species, which consists of groups of strains that differ from each other by the presence of some genetic markers, mainly plasmids and regulatory genes [2,3,4]. For B. anthracis, these determinants are primarily two plasmids: pXO1 and pXO2. pXO2 harbors genes (capBCAD operon) that encode enzymes for the synthesis of a capsule consisting of poly-D-γ-glutamic acid. This capsule covers the surface of the B. anthracis vegetative cell and protects it from host immune responses [5]. pXO1 harbors the genes of the tripartite anthrax toxin complex components: protective antigen (PA, encoded by pagA), lethal factor (LF, encoded by lef), and edematous factor (EF encoded by cya). PA is a pore-forming protein that forms complexes with LF or EF, called lethal toxin (LT) and edematous toxin (ET), respectively, and allows the effector proteins LF or EF to enter the cytosol of mammalian cells. LF is a Zn-dependent metalloproteinase that is specific for mitogen-activated kinases (MAPKKs or MEKs). The LF-caused cleavage of MAPKKs disrupts the signaling pathways of the host cells. EF is a calmodulin-dependent adenylate cyclase, which also disrupts the signaling pathways of host cells by increasing the level of synthesis of cyclic adenosine monophosphate (cAMP). Clinically, the effect of EF is manifested as tissue edema, which served as the basis for the name of this protein [6,7,8]. The emergence of these pathogenicity factors required a change in the protein expression regulation system. In B. anthracis, this happened through two genetic events: first, the inactivation of the global transcriptional regulator PlcR via a point nonsense mutation, which regulates, among other things, the expression of the pathogenicity factors of other B. cereus complex species; and second, the acquisition of a new transcriptional regulator AtxA. The gene atxA is located on the pXO1 plasmid. AtxA, directly and indirectly, controls the expression of more than a hundred genes localized on the chromosome and on both pathogenicity plasmids, including genes for capsule and toxin synthesis [9,10]. AtxA is critically important for the anthrax microbe to realize its pathogenic potential. Like the toxin and capsule, AtxA could be called a pathogenicity factor.
pXO1 and pXO2 are traditionally considered specific for B. anthracis; nevertheless, some B. cereus strains that possess pXO-like plasmids have recently been discovered. Some strains were identified as a separate phylogenetic unit: B. cereus biovar anthracis [11,12,13]. B. cereus biovar anthracis and B. cereus possessing pXO-like plasmids still differ from the anthrax microbe primarily in their pathogenicity. Such strains seem to be less pathogenic for humans than B. anthracis, as only a few clinical cases have been described [13]. However, at the same time, they are virulent for other large animals, including great apes and laboratory animals [11,12,13].
Acquisition of the pathogenicity factors described above and formation of the B. anthracis species occurred, according to modern estimations, 12–26 thousand years ago [11,14], so the global population of the anthrax microbe did not have time to divide into subspecies or any other small groups that would differ phenotypically. Nevertheless, several molecular typing methods make it possible to distinguish genetically different groups of strains. One of the most widely used methods for dividing the B. anthracis species into phylogenetic groups is typing based on a small set of so-called “canonical” single nucleotide polymorphisms (canSNP). B. anthracis strains are divided among canSNP-groups into three main evolutionary lines: A, B, and C [15].
The A lineage is the most genetically diverse; it includes the canSNP groups A.Br.008/009, A.Br.001/002, A.Br.Ames, A.Br.003/004, A.Br.005/006, A.Br.Aust94, A.Br.Vollum, and A.Br.WNA. Strains of these groups were found worldwide, but some of their geographical distribution patterns can be distinguished [16,17]. The most widespread group is the canSNP A.Br.008/009 group, also referred to as Trans-Eurasian (TEA), the strains of which circulate in Eurasia and are very widely spread in Russia. This group is divided into two subgroups: A.Br.008/011 and A.Br.011/009 [18]. Group A.Br.001/002 is widespread in China (central and eastern regions) and is the ancestor of the small group A.Br.Ames, which was brought to the United States from China [19].
Groups A.Br.WNA and A.Br.003/004 are widely represented in North and South America; group A.Br.005/006 is found in Australia, Africa, and Europe; group A.Br.Aust94 is distributed in Southeast Asia, India, Australia, western China, and Turkey; and the A.Br.Vollum group is spread across South Africa and some regions of Asia (Pakistan and Afghanistan) [16]. The B lineage is more limited in its distribution area. It includes the canSNP group B.Br.CNEVA, found in Central Europe; B.Br.Kruger, located in South Africa; and B.Br.001/002, the strains of which are spread across South Africa, Europe, and Korea, and which were recently also found in the Russian Arctic [16,20]. The C lineage includes only the canSNP group C.Br.001, which is occasionally found in North America. It is believed that this lineage evolved the slowest of all; therefore, it is the most genetically close to the putative common ancestor of all B. anthracis lineages and the most distant from the rest of the anthrax microbe population [15,21].
The C lineage is distinguished by having the lowest evolutionary rate, the most significant degree of genetic difference from the rest of the populations of the anthrax microbe, and being the closest to the putative common ancestor of all B. anthracis lineages [15,21].
Such a scheme of B. anthracis global population genotyping is quite convenient. However, it is not entirely exhaustive. Therefore, additional SNPs are often proposed for a more detailed phylogenetic analysis, making it possible to isolate new phylogenetic and phylogeographic groups [22,23]. However, mutations in the genes encoding pathogenicity factors have practically not been used before as additional markers for genotyping. Moreover, unlike many other pathogens [24,25,26,27,28,29], the allelic polymorphism of these genes in the anthrax microbe is practically not described. Only a few articles have been published in which the allelic polymorphism of individual pathogenicity genes was assessed among a small number of strains [30].
In this work, we attempted to describe the allelic polymorphism of pagA, lef, cya, and atxA genes among the B. anthracis strains from our collection and to analyze the data obtained in terms of their correlation with strains belonging to a particular evolutionary lineage and canSNP group, as well as with the geographic origin of these strains.
2. Materials and Methods
2.1. Strains and Growth Condition
In this work, we used 40 live B. anthracis strains from the SRCAMB collection listed in Table 1 (State Research Center for Applied Microbiology and Biotechnology, Obolensk, Russia). All these strains were isolated in the territory of Russia and the former USSR. Vegetative cells of B.anthracis strains were cultured on BHI agar (SRCAMB), then inactivated, and sterile total genomic DNA was isolated using the Genomic DNA Purification Kit (Thermo Fisher Scientific, MA, USA). All manipulations were performed in a biosafety laboratory level 3. The sterility of the DNA samples was confirmed by cultivating 5% of the final DNA volume with negative results. The DNA concentration was quantified using the NanoDrop Onec spectrophotometer (Thermo Fisher Scientific).

Table 1.
Strains used in this work.
Additionally, we used the whole-genome sequencing data deposited in GenBank for 48 strains of B. anthracis and B. cereus.
2.2. Whole-Genome Sequencing
DNA libraries were prepared using the Nextera DNA Library Preparation Kit (Albiogen, Moscow, Russia). Whole-genome sequencing was performed using the Illumina MiSeq and Ion Torrent PGM instruments and the corresponding reagent kits: Ion PGM Reagents 400 Kit, Ion 318 Chip Kit (Life Technologies, Moscow, Russia), and MiSeq Reagent Kit v3 (Albiogen, Moscow, Russia).
2.3. Data Analysis
The assembling of the pXO1 plasmid sequence was performed using the DNASTAR software package (Lasergene, Madison, WI, USA) (https://www.dnastar.com/ perpetual license, accessed on 1 November 2021). The genome of the B. anthracis Ames Ancestor strain (plasmid pXO1, GenBank: AE017336.2) was used as a reference genome. The identified mutations and their coordinates were described in accordance with the reference genome. The in silico translation of nucleotide sequences into amino acid sequences was performed using the Transeq (EMBOSS, https://www.ebi.ac.uk/Tools/st/, accessed on 1 November 2021) The multiple alignments of nucleotide sequences were performed using the MEGA 7.0 software package (http://www.megasoftware.net, accessed on 1 November 2021). Simpson’s diversity index D was calculated using the PHYLOViZ 2.0 software [31]. Phylogenetic analysis was performed using the MEGA 7.0 (for UPGMA) and PHYLOViZ 2.0 (for goeBURST) software.
3. Results
Among the studied samples, we identified 11 alleles (sequence types, STs) of the gene pagA (D = 0.7717 (0.7231 ÷ 0.8202)), nine alleles each from the genes lef (D = 0.6468 (0.5585 ÷ 0.7352)) and cya (D = 0.6358 (0.5452 ÷ 0.7265)), and two alleles of the gene atxA (D = 0.023 (–0.0214 ÷ 0.0669)). All the STs differed from each other only by their single-nucleotide substitutions; we failed to find any deletions, insertions, or even substitutions of two nucleotides in a row. The identified STs, indicating their differences from the ST of the reference genome, are listed in Table 2, Table 3 and Table 4. Each gene ST to which the reference Ames Ancestor strain belonged was designated as ST1, then numbering was carried out in order of the decreasing number of strains of B. anthracis, after which STs, including strains of B. cereus, were numbered.
Table 2.
Sequence types of the gene pagA.
Table 3.
Sequence types of the gene lef.
Table 4.
Sequence types of the gene cya.
To assess the phenotypic manifestation of the identified nucleotide polymorphism—that is, whether the nucleotide substitution in each identified position is synonymous, leads to an amino acid substitution of the corresponding protein, or leads to its inactivation due to the appearance of a stop codon—we performed the in silico translation of nucleotide sequences into amino acid sequences. The coordinates of the predicted amino acid substitutions, indicating in which protein domain each substitution occurred, are shown in Table 5, Table 6 and Table 7. In these tables, the coordinates of amino acid substitutions are indicated for the complete sequence of translated proteins, excluding their post-translational modification, which consisted of the cleavage of the N-terminal signal sequence. In the PA protein, this is a 29 amino acid sequence; therefore, the mature protein length was reduced from 764 to 735 amino acids [32]. In LF and EF, this is a 33 amino acid sequence, so their length was reduced from 808 to 776 [33] and from 800 to 767 amino acids [34], respectively.
Table 5.
Predicted amino acid substitution in the PA protein.
Table 6.
Predicted amino acid substitution in the LF protein.
Table 7.
Predicted amino acid substitution in the EF protein.
The tables do not show data for the atxA, since there is practically no polymorphism for this gene. Even the B. cereus strains 03BB102 and G9241, significantly different from B. anthracis strains, carry the same allele of atxA as the rest of the sample. The only mutation of this gene was found in the B. cereus biovar anthracis strain CI. The single-nucleotide mutation atxA 563T→A leads to the AtxA 188I→N amino acid substitution. Nevertheless, despite the extremely low degree of atxA, we did not exclude it from further work and used SNP atxA 563T→A as a marker, the presence of which emphasizes the genetic remoteness of the CI strain from the rest of the studied sample.
The division of the studied strains into genotypes (GT) based on a certain combination of studied genes STs is shown in Table 8. GTs were numbered according to the following principle; GT, which included the reference strain, was designated as GT1, and subsequent GTs were numbered in decreasing order of the number of strains in which they were detected, with the last numbers being assigned to the GTs of B. cereus strains. In total, we identified 19 genotypes (D = 0,8908 (0,8604 ÷ 0,9212)). More details on GTs are given in Table S1.
Table 8.
Separation of the studied strains into genotypes based on the pagA, lef, cya, and atxA ST combination.
By describing the allelic polymorphism of pathogenicity genes localized on the pXO1 plasmid, identifying their sequence types, and dividing the strains of the studied sample by genotypes based on a certain set of sequence types of individual genes, we thus applied the MVLST method (Multi-Virulence Locus Sequence Typing).
In the present work, we limited our study to only the genes located on the pXO1 plasmid, not taking into account the genes of capsule synthesis; therefore, this molecular typing scheme can be designated as MVLSTpXO1 genotyping, and individual genotypes can be designated as MVLSTpXO1 GTs. It is noteworthy that the division of strains into the MVLSTpXO1 GT, in general, repeats its division into canSNP groups. Even the diversity index is very similar—0,8908 (0,8604 ÷ 0,9212) for MVLSTpXO1 and 0,8582 (0,8037 ÷ 0,9126) for canSNP typing. However, this observed pattern was not absolute; in some cases, strains of several canSNP groups formed a common GT, or, on the contrary, strains of one canSNP group were split into several GTs. Below, we provide a description of such exceptions.
GT1 includes 11 strains: 7 out of 12 strains of the canSNP group A.Br.001/002 and all four strains of the group A.Br.Ames. Five strains of the A.Br.001/002 group, not included in GT1, form GT7, which is distinguished by one nonsynonymous SNP lef 895G→T (LF299A→S).
GT2 includes 18 strains: 15 out of 32 strains of the line TEA (12 strains of the A.Br.008/011 canSNP group and three strains of the canSNP group A.Br.011/009), both strains of the A.Br.003/004 group, and one of the five strains of the group A.Br.005/006. All strains of the canSNP group A.Br.008/011 not included in GT2 (n = 17) form GT3, which differs from GT2 in two SNPs: pagA 981A→T and lef 2126A→G. Four strains of the A.Br.005/006 group, not included in GT2, form two separate genotypes: GT8 (n = 3), which has an additional unique non-synonymous SNP cya 2129A→C (EF 710H→P), and GT11 (n = 1), which lacks an SNP cya 600C→T specific for GT2.
GT5 includes seven strains: all five strains of the B.Br.CNEVA group isolated in Central Europe (except strain 44, whose place of isolation is unknown) and two strains of the B.Br.001/002 group isolated in the Russian Arctic. This genotype did not include the remaining five strains of the B.Br.001/002 group from the studied sample. Three of them were isolated in Siberia, the Baltic region, and Korea from GT9, which differs from GT5 by its additional unique nonsynonymous SNP pagA 1297A→G. Two other strains isolated in Sweden and South Africa form the separate GT10 due to the absence of SNP pagA 1799C→T. This SNP is also absent in the Kruger B strain, the only representative of the Br.Kruger group (GT15) in our sample. Instead of this SNP, GT15 has a unique SNP pagA 1765C→A.
Strains of the A.Br.Aust94 group also form two genotypes: seven of its eight strains were included in GT4, and the Ohio ACB strain is allocated into a separate genotype GT14 due to the presence of the unique synonymous SNP pagA 1803T→C.
The phylogenetic relationships of all the MVLSTpX1 genotypes described in this work are illustrated in Figure 1. As can be seen from this figure, and which is quite expected in view of the above, MVLSTpXO1 genotypes form clusters belonging to different evolutionary lines: A, B, and C. The most numerous «A» cluster is divided into two subclusters. The first of them combines GT1, GT4, GT7, and GT14—that is, strains of canSNP groups A.Br.001/002, A.Br.Ames, and A.Br.Aust94. The second one includes GT2, GT3, GT6, GT8, GT11, GT12, and GT13—i.e., strains of the canSNP groups A.Br.003/004, A.Br.005/006, A.Br.005/007, A.Br.Vollum, A.Br.WNA, and A.Br.008/009 (subgroups A.Br.008/011 and A.Br.011/009). GT16, which includes the only strain of C-lineage, is far from clusters A and B and, in fact, occupies an intermediate position between B. anthracis and B. cereus strains. B. cereus biovar anthracis is the most distant from the rest of the studied sample.
Figure 1.
UPGMA dendrogram illustrating the phylogenetic relationships of MVLSTpXO1 genotypes. Opposite each GT, it is indicated in parentheses which canSNP groups this GT includes. For each GT, which includes strains that do not belong to B. anthracis, the species is indicated instead of the canSNP group. Braces and colors mark genotypes belonging to the different evolutionary lineages: A (red color), B (blue color), and C (green color).
Additionally, we carried out phylogenetic analysis using the goeBURST algorithm [35] (Figure 2). As a result, we found that GT2 is linked simultaneously with nine genotypes: GT3, GT4, GT5, GT6, GT8, GT11, GT12, GT13, and GT16. This GT2 differs from GT1 by three SNP: pagA 195 C→T, pagA 1799 C→T, and cya 600C→T. All three SNPs are characteristic for most of the strains in our sample: synonymous SNP pagA 195C→T is found in 64 strains, SNP pagA 1799C→T (PA 600A→V) is found in 61 strains, and all of them also possess the SNP pagA 195C→T. The synonymous SNP cya 600C→T is found in 71 strains. All three SNPs together are found in 60 strains, but 42 of them have additional SNPs, due to which some other GTs are formed. Thus, we can assume that for the studied sample, GT2 with its three characteristic SNPs pagA 195 C→T, pagA 1799C→T, and cya 600C→T is the most typical; other GTs either lack these markers or acquire additional SNPs. In goeBURST analysis, all genotypes form three main clonal complexes (CC) unite genotypes, the genetic distance between which is minimal and equal to one (Figure 2). Two of them include some GTs of A lineage. The first one remains identical to the corresponding subcluster on the dendrogram shown in Figure 1 and includes GT1, GT4, GT7, and GT14—that is, canSNP groups A.Br.001/002, A.Br.Ames, and A.Br.Aust94. The second CC is formed by GT2, GT6, GT8, and GT11—that is, by the groups A.Br.003/004, A.Br.005/006, and A.Br.Vollum and the strains of the TEA line (groups A.Br. 008/011 and A.Br.011 / 009) lacking the SNP pagA 981A→T. The third CC is the most genetically distant from the first two and unites all genotypes of the B lineage (groups B.Br.001/002, B.Br.CNEVA, and B.Br.Kruger).
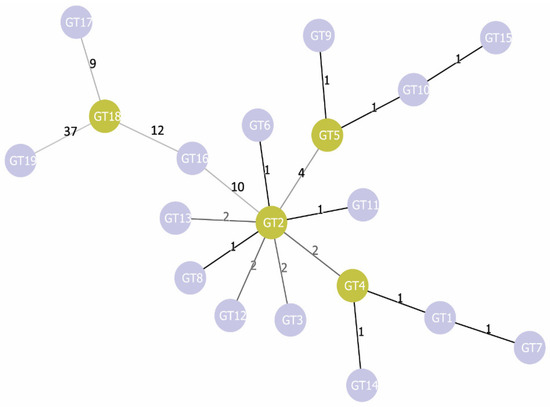
Figure 2.
Phylogenetic tree of MVLSTpXO1 genotypes constructed using the goeBURST algorithm. The numbers above the line linking two genotypes indicate the genetic distance between these genotypes.
4. Discussion
As is known, anthrax mainly affects ungulates [1], including horses and livestock—that is, animals that are still the basis of agriculture, and until modern times were also the only land transport. As a result, B. anthracis could easily be introduced into different regions not only by the natural migrations of ungulates but also through human activities, such as colonization, military operations, and trade expeditions. In the course of such activities, not only did people themselves move, but so too did triding and sled animals; pack animals; livestock; and livestock products, such as leather, fur, woolen clothing, and harnesses, as well as items of ammunition. These animals could be sick with anthrax, and goods could be contaminated with B. anthracis spores, which are easily transported and remain viable. Perhaps the anthropogenic transportation of spores was the main driver of the spread of B. anthracis across the planet [14]. Due to the natural and anthropogenic transfer, genetically related strains may be introduced into distant regions, where their further evolution may proceed differently. At the same time, genetically distinct strains from different origins can be brought into the same area over and over again. As a result, local populations of the anthrax microbe can be represented by strains of different genetic lines. Therefore, the geographical origin of a particular B. anthracis strain is not very informative in itself, but rather should be considered in conjunction with its genetic profile, as well as the natural, historical, and economic characteristics of the region where this strain was isolated. Based on this, we attempted to identify any correlations between the geographical origin of the strains from our sample, their canSNP groups, and their MVLSTpXO1 genotypes.
In this analysis, the first thing that caught our attention was the synonymous SNP pagA 981A→T. This SNP had earlier been proposed as a specific marker for a small subgroup of the canSNP group A.Br.008/009, which was originally isolated in Russia (the so-called Sverdlovsk group) but has also been found in Norway, Hungary, and Slovakia [36]. A little later, Eremenko, during the genotyping of B. anthracis strains from the Stavropol Antiplague Institute collection, found this SNP in all eight strains belonging to the canSNP group A.Br.008/009 in the studied sample [37]. In the present study, we found SNP pagA 981A→T in 17 out of the 27 strains in the A.Br.008/011 group (as we have already indicated, A.Br.008/011 together with group A.Br.011/009 is included in the A.Br.008/009 group) isolated in the territory of the former USSR (Table 1, Table 8 and Table S1). Still, we did not find it in any strain from the comparison group—that is, isolated in the other regions. All 17 strains belong to MVLSTpXO1 GT-3 (Table 8 and Table S1)—i.e., they have no other differences among themselves in terms of the sequence of genes described in this work. Thus, our results complement the data of Eremenko and Okinaka [36,37] and indicate that SNP pagA 981A→T is a marker of a rather large subgroup of the A.Br.008/011 group. This subgroup is spread over territory stretching over 6000 km from west to east and about 4000 km from north to south, including Central Asia, the Caucasus, the Black Sea and Caspian steppes, the European part of Russia, and even the eastern Arctic (Yakutia). Moreover, the A.Br.008/011 pagA 981A→T subgroup (or MVLSTpXO1 GT-3) includes, if not the majority, then at least half of the strains of the A.Br.008/011 group circulating in the territory of the former USSR and the former Russian Empire. Additionally, from a deeper historical perspective, the GT3 strain distribution area coincides with the territories that, in the XII–XVIII centuries, were part of the Mongol Empire and the states into which it disintegrated (Golden Horde, Chagatai Horde, Ilkhanat), as well as the zones of their political influence (Novgorod region of Russia) and territories subjected to Mongol military campaigns (Hungary and Slovakia). Earlier, we hypothesized about the significant role of the Mongol invasion of the west in the spread of anthrax across Eurasia [20]. The geographical area of distribution of the subgroup A.Br.008/011 pagA 981A→T described in this article additionally supports our hypothesis. Even the fact that one of the strains of this subgroup was isolated in Norway fits well into the framework of this concept. The northeast region of Russia—Novgorod and Pskov principalities—through most of its history was associated with Scandinavian states not only by trade ties but also by a non-stop series of military conflicts, which may have led to the introduction of A.Br.008/011 pagA 981A→T strains into Scandinavia through the exchange of goods and/or war booty.
As mentioned above, in addition to pagA 981A→T we found several more SNPs dividing the strains of one canSNP group into different GTs. Additionally, there is some reason to link these GTs to a specific geographic region.
For example, the unique SNP pagA 1803T→C distinguishes strain Ohio ACB from other strains of the canSNP group A.Br.Aust94 in our sample. Additionally, this is the only strain isolated in the USA, while other strains of the group A.Br.Aust94 were isolated in the Old World. This data prompted us to evaluate the possibility of using SNP pagA 1803T→C to determine the origin of the strain of the A.Br.Aust94 group from North America. Not having such strains in our collection, we had to limit ourselves to a small number of deposited archives with the raw WGS data of the A.Br.Aust94 group isolated in the USA (Table 9). However, contrary to our hopes, we found this SNP in only two American strains out of ten. Therefore, the maximum that can be said about the diagnostic role of this SNP is that it occurs in some A.Br.Aust94 strains circulating in the United States.
Table 9.
Presence of SNP pagA 1803T→C in strains of the A.Br.Aust94 group of American origin.
Another exciting marker is SNP lef 895G→T, due to the presence of which five of the twelve strains of canSNP group A.Br.001/002 form a separate genotype GT7, while most of the strains of this group are included in GT1. We were interested in the fact that the strains with this SNP were isolated mainly in the New World—the USA, Brazil, and Jamaica—with a single one being isolated in Bangladesh. Strains lacking this SNP were isolated in Russia, China, Kazakhstan, and Germany. To find out whether the SNP lef895G→T is characteristic of strains of the A. Br.001/002 group of specifically American origin, we analyzed 42 additional genomes of B. anthracis strains of this group, of which three strains were of American origin, 28 were of European origin, and 11 strains were of Asian origin. We found this SNP in all three American strains, but also in 23 of 28 European and 8 of 11 Asian strains (Table 10).
Table 10.
Presence of SNP lef 895G→T in strains of the A.Br.001/002 group of different geographical origin.
Thus, SNP lef 895G→T was found to be typical for most strains of the A.Br.001/002 group, regardless of their geographic origin. Additionally, we can make some inferences about the absence of this SNP as a marker of a subgroup of the A.Br.001/002 group. This subgroup has the same GT as the A.Br.Ames group, therefore, is more related to it (within the framework of the typing scheme used). Considering that the A.Br.Ames group is descended from the A.Br.001/002 group [19], it can be assumed that the GT1 subgroup of the A.Br.001/002 group is a kind of “transitional link” that has already acquired plasmid markers of the A.Br.Ames group, which has not yet developed the chromosomal canSNPs of this group.
Unfortunately, unlike the A.Br.008/011 pagA 981A→T subgroup, we do not have data that could somehow explain the reasons behind and patterns of distribution of the A.Br.Aust94 pagA 1803T→C and A.Br.001/002 lef 895G→T subgroups.
Interestingly, lef 895 is the only position that we found that possesses three allelic states—G, T, and A. lef 895G is found in most strains; lef 895A is a marker found in all canSNP strains of lineage B canSNP groups; and lef 895T, as indicated above, is a specific marker of the GT7 subgroup of A.Br.001/002 strains. However, besides the group of A.Br.001/002 strains, lef 895T is unexpectedly found in a non-anthracis strain, B. cereus G924.
Moreover, we would like to focus on lineage B strains. Previously it was shown that B. anthracis strains of the B.Br.001/002 group circulating in Siberia form a separate phylogenetic cluster, named «Siberia» [38]. However, in our work two Siberian B.Br.001/002 strains were combined into one GT5 with the B.Br.CNEVA group, but not with other B.Br.001/002 strains, including not with the strain I-364 isolated in Siberia. Both of these strains were isolated in the Arctic. Strain Yamal-2 was isolated during an anthrax outbreak in Yamal in 2016, which occurred due to the thawing of soil anthrax conserved in permafrost. Strain LP53/5YA was found in permafrost during paleontological excavations in Yakutia. The very history of these strains, such as their isolation in the Arctic region, including isolation from the permafrost, indirectly indicates that their age is most likely older than that of other strains of this line. In this case, GT5 unites the B.Br.CNEVA group and the archaic strains of the B.Br.001/002 group and this GT was formed before the evolutionary divergence of these two canSNP groups. Moreover, the fact of the certain phylogenetic closeness of strains circulating in Central Europe with strains isolated in the Arctic, which is remote from Europe and is not connected to it by animal migration routes or economic ties, remains intriguing.
The similarity of strains from the Arctic and Central Europe raises questions not only about the past of these strains (how exactly and when they spread between these regions and within them) but also about their future. Global climate changes with a certain probability can lead to the melting of permafrost, and thawing of soil foci of anthrax, as it happened in 2016 in Yamal. Moreover, the random nature of the finding of Yakut strains illustrates that viable spores of Bacillus anthracis can be distributed in permafrost much more widely than it seems at the moment. A change of permafrost boundaries means not only the de-preservation of the microorganisms presented in it but also means an improvement of the conditions for the plants’ vegetation. In turn, this means an increase in the food supply of animals, which can potentially lead to a rise in the number of endemic animals and migrations of animals to the region from low-latitude areas. That would also create favorable conditions for increasing the livestock of farm animals, primarily deer, which are traditionally bred in a nomadic way, that is, the maximum number which directly depends on the productivity of pastures. Such an increase in the number of animals is likely to outpace the development of the region’s infrastructure, including medical and veterinary supervision, which could provide timely prevention of anthrax outbreaks. In epidemiological investigations of such episodes, any similarity of the Arctic strains with the Central European strains can play a cruel joke and lead to the mistaken impression that the strain that caused the outbreak is not endemic but brought from Europe.
In addition to describing the distribution of certain markers in phylogenetic and phylogeographic groups of B. anthracis, it would be interesting to try to find relationships between the MVLSTpXO1 genotype of such groups and its phenotypic manifestation. Since the function of the genes used in this genotyping scheme is the synthesis of toxins that directly affect the host organism, such a phenotypic manifestation could be differences in the pathogenicity of strains with different MVLSTpXO1 genotypes. This approach is more or less applicable to other pathogens [24,25,26], but it is very difficult for the anthrax microbe.
It is hardly possible to identify differences in pathogenicity in natural conditions during anthrax outbreaks—due to modern sanitary and veterinary control, such outbreaks are eliminated by antibiotic therapy, the vaccination of at-risk groups, and the slaughter of sick animals before statistically reliable data can be collected. Therefore, any assumptions that we can make about the potential influence of the MVLSTpXO1 genotype are highly speculative and do not have proper experimental confirmation. Nevertheless, we consider it possible to state a number of such suggestions.
The first thing that can be noted is the presence of four vaccine strains in the studied sample—A16R, obtained in the 1950s in China [39]; Sterne, obtained in South Africa in the 1930s, which is widely used as a veterinary vaccine throughout the world [40]; V770-NP-1R, which has been used as a producer for the AVA vaccine in the United States since the 1970s; and STI-1, which has been used as a live vaccine for humans since the 1940s in the USSR and now in Russia. Thus, the sample includes the most widespread vaccine strains, obtained independently in different parts of the world. All four of these strains retained the pXO1 plasmid but lost the pXO2 plasmid. Our analysis shows that none of these strains acquired any peculiarities in the sequence of toxin synthesis genes that distinguish them from natural, completely virulent strains. Moreover, the A16R strain does not differ in terms of the sequence of these genes from its parent strain A16. Thus, if the attenuation of B. anthracis strains is caused by any changes in the genome, in addition to the loss of the capsule formation plasmid [41], then these changes do not affect the genes involved in toxin synthesis.
The second point regarding the comparative virulence of B. anthracis strains that we would like to discuss concerns the modeling of anthrax. Although, as we indicated above, it is hardly possible to reveal such facts using laboratory models, we found in the literature a description of one exception to this pattern. When guinea pigs were vaccinated with the AVA vaccine, it was found that during subsequent infection different strains of B. anthracis overcome post-vaccination immunity with different efficacies—the survival rate of animals varied from 6 to 100% [42]. The authors could not find a correlation of this feature of the strains with any other characteristics, including their geographical origin. However, here it is worth paying attention to the composition of AVA; it is a precipitate of the culture fluid of the nonencapsulated strain V770-NP1-R (belongs to A.Br.003/004 and GT2)—that is, the PA, LF, and EF of this strain act as immunogenic determinants [43]. If the LF, EF, and PA sequences of the infecting strain and the V770-NP1-R strain are different, then the antibodies generated after vaccination may poorly recognize the toxins of the infecting strain, which may lead to an increase in the death rate of the infected animals. In this case, the ability of a strain to overcome post-vaccination immunity may depend more on its toxin synthesis gene sequence—that is, the MVLSTpXO1 genotype—than on other characteristics, including the region where the strain was isolated. Unfortunately, we could not verify this assumption, since we could not find in the available sources any data that could be used to determine the MVLSTpXO1 of the strains used in [42]—the WGS data or at least their canSNP group, which, as we have shown in the present article, correlates with MVLSTpXO1 genotype.
However, whatever the interesting data obtained in the laboratory experiments are, they cannot fully reflect the natural virulence of the B. anthracis strains. For evaluating the hypothesis concerning the influence of the MVLSTpXO1 genotype on the strain pathogenicity, the evidence collected during anthrax outbreaks among naturally susceptible animals (ungulates) is more important. We managed to find the results of an epidemiological analysis in the literature, which could be interpreted as indirect evidence of our hypothesis. When comparing two anthrax-endemic South African ecosystems—Etosha National Parks (Namibia) and Kruger National Parks (Republic of South Africa), which are home to large populations of zebras and Kudu—it was noted that in Etosha Park almost half of all reported cases of anthrax occur in zebras, while the share occurring in kudu is less than 1%. In Kruger Park, a diametrically opposite picture was observed—more than half of the cases were recorded in kudu, while zebras get sick far more rarely [1,44,45]. These two regions differ in terms of the biodiversity of the B. anthracis strains circulating there. In Etosha Park, strains of the A.Br.Aust94 group prevails, while strains of the A.Br.008/009 group are rarely found. Outside the park, in the territory of Namibia, strains of groups A.Br.005/006 and A.Br.001/002 have also been identified, while strains of B lineage have not been found in this country [46]. In the Republic of South Africa, the population of the anthrax microbe is more diverse; there are circulating B. anthracis strains of both A and B lineages belonging to the canSNP groups A.Br.003/004, A.Br.005/006, A.Br.Aust94, B.Br.Kruger, and B.Br.001/002 [47], while strains of B lineage (B.Br.Kruger and B.Br.001 002) were isolated directly in Kruger Park. This biodiversity is even used as an argument in favor of the South African origin of the B. anthracis species, although, in our opinion, it is just as likely to be due to the political and economic history of this region. South Africa has experienced a number of events, each of which could potentially be associated with the introduction of different strains of B. anthracis into the region through livestock and animal products. These include the migration of the Bantu pastoralist tribes from the north; two waves of European colonization from the Dutch (Boers) and the British; and two Anglo-Boer wars, in which riding and pack animals, supplies, and equipment for the warring parties were supplied from different countries. In addition, South Africa, due to its geographical position has been a major transit point for all European trade with the Indian Ocean region from the XVII century until the opening of navigation through the Suez Canal. In addition, it was South Africa, due to its geographical position, that was the main transit point for all European trade with the Indian Ocean basin from the 17th century until the opening of navigation along the Suez Canal. Such active movements of large masses of people, livestock, and goods, including livestock products, over several centuries, could lead to the contrasting high genetic diversity of B. anthracis strains in this region. Strains of the B lineage are currently circulating in South Africa. It could be assumed that these strains are capable of infecting different ungulates that differ evolutionarily and physiologically with greater efficiency than strains of line A, whether they are artiodactyl ruminants bovids (kudu) or non-artiodactyl monogastric equines (zebras). As one of the reasons for such a potential difference in virulence, we could suggest differences in the sequence of anthrax toxins in strains A and B, as shown in this article.
We are aware that our assumptions are too speculative and can hardly be tested in practice. If there is a difference in the manifestation of pathogenic properties in strains with the MVLSTpXO1 genotype, then it is unlikely that it has a tangible epidemiological significance. Nevertheless, MVLST-typing, which primarily reflects the evolution of the main effector proteins, the acquisition of which turned B. anthracis into a dangerous pathogen, is of interest because at least in some cases it makes it possible to distinguish geographically separate groups of strains within the canSNP groups. The use of this typing method, especially in combination with other more common genotyping schemes, can to some extent bring us closer to a more complete understanding of the evolutionary patterns of the anthrax microbe and the distribution of its individual intraspecific groups across the globe.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/pathogens10121556/s1, Table S1. Distribution of the studied strains by the MVLST-pXO1 genotypes (MVLST-pXO1-GT) depending on the combination of pagA, lef, cya, and atxA gene sequence types (ST).
Author Contributions
The authors contributed as follows: conceptualization, V.T.; methodology, G.T., T.K. and I.B.; validation, I.B.; formal analysis, Y.G. and V.T.; investigation, Y.G., G.T. and T.K.; resources, I.D.; data curation, V.T. and I.B.; writing—original draft preparation, V.T., Y.G. and A.L.; visualization, I.B.; supervision, V.T.; project administration, I.D. and V.T.; funding acquisition, I.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation, agreement No. 075-15.2019-1671 dated 31 October 2019. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used for this study are available in the text of the article and in the Supplementary Materials. Additional data including generated genomic sequences of this study are available from the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor) but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor).
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Chapter 3: Anthrax in animals. In Anthrax in Humans and Animals, 4th ed.; World Health Organization: Geneva, Switzerland, 2008; Available online: https://www.ncbi.nlm.nih.gov/books/NBK310481/ (accessed on 1 November 2021).
- Ehling-Schulz, M.; Lereclus, D.; Koehler, T.M. The Bacillus cereus Group: Bacillus Species with Pathogenic Potential. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Helgason, E.; Okstad, O.A.; Caugan, D.A.; Johansen, H.A.; Fouet, A.; Mock, M.; Hegna, I.; Kolstø, A.B. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis—One species on the basis of genetic evidence. Appl. Environ. Microbiol. 2000, 66, 2627–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasko, D.A.; Altherr, M.R.; Han, C.S.; Ravel, J. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol. Rev. 2005, 29, 303–329. [Google Scholar] [CrossRef]
- Sharma, S.; Bhatnagar, R.; Gaur, D. Bacillus anthracis Poly-γ-D-Glutamate Capsule Inhibits Opsonic Phagocytosis by Impeding Complement Activation. Front. Immunol. 2020, 11, 462. [Google Scholar] [CrossRef]
- Collier, R.J. Membrane translocation by anthrax toxin. Mol. Asp. Med. 2009, 30, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swick, M.C.; Koehler, T.M.; Driks, A. Surviving Between Hosts: Sporulation and Transmission. Microbiol. Spectr. 2016, 4, 529–566. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar] [CrossRef] [Green Version]
- Fouet, A. AtxA, a Bacillus anthracis global virulence regulator. Res. Microbiol. 2010, 161, 735–742. [Google Scholar] [CrossRef]
- McCall, R.M.; Sievers, M.E.; Fattah, R.; Ghirlando, R.; Pomerantsev, A.P.; Leppla, S.H. Bacillus anthracis Virulence Regulator AtxA Binds Specifically to the pagA Promoter Region. J. Bacteriol. 2019, 201, e00569-19. [Google Scholar] [CrossRef]
- Kolstø, A.B.; Tourasse, N.J.; Okstad, O.A. What sets Bacillus anthracis apart from other Bacillus species? Annu. Rev. Microbiol. 2009, 63, 451–476. [Google Scholar] [CrossRef]
- Antonation, K.S.; Grützmacher, K.; Dupke, S.; Mabon, P.; Zimmermann, F.; Lankester, F.; Peller, T.; Feistner, A.; Todd, A.; Herbinger, I.; et al. Bacillus cereus Biovar Anthracis Causing Anthrax in Sub-Saharan Africa-Chromosomal Monophyly and Broad Geographic Distribution. PLoS Negl. Trop. Dis. 2016, 10, e0004923. [Google Scholar] [CrossRef]
- Hoffmaster, A.R.; Hill, K.K.; Gee, J.E.; Marston, C.K.; De, B.K.; Popovic, T.; Sue, D.; Wilkins, P.P.; Avashia, S.B.; Drumgoole, R.; et al. Characterization of Bacillus cereus isolates associated with fatal pneumonias: Strains are closely related to Bacillus anthracis and harbor B. anthracis virulence genes. J. Clin. Microbiol. 2006, 44, 3352–3360. [Google Scholar] [CrossRef] [Green Version]
- Vergnaud, G. Bacillus anthracis Evolution: Taking Advantage of the Topology of the Phylogenetic Tree and Human History to Propose Dating Points. Erciyes Med. J. 2020, 42, 362–369. [Google Scholar]
- Van Ert, M.N.; Easterday, W.R.; Huynh, L.Y.; Okinaka, R.T.; Hugh-Jones, M.E.; Ravel, J.; Zanecki, S.R.; Pearson, T.; Simonson, T.S.; U’Ren, J.M.; et al. Global genetic population structure of Bacillus anthracis. PLoS ONE 2007, 2, e461. [Google Scholar] [CrossRef] [Green Version]
- Derzelle, S.; Thierry, S. Genetic Diversity of Bacillus anthracis in Europe: Genotyping Methods in Forensic and Epidemiologic Investigations. Biosecur. Bioterror. 2013, 11, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Pilo, P.; Frey, J. Pathogenicity, population genetics and dissemination of Bacillus anthracis. Infect. Genet. Evol. 2018, 64, 115–125. [Google Scholar] [CrossRef]
- Chiaverini, A.; Abdel-Glil, M.Y.; Linde, J.; Galante, D.; Rondinone, V.; Fasanella, A.; Cammà, C.; D’Alterio, N.; Garofolo, G.; Tomaso, H. Whole Genome Sequencing for Studying Bacillus anthracis from an Outbreak in the Abruzzo Region of Italy. Microorganisms 2020, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Simonson, T.S.; Okinaka, R.T.; Wang, B.; Easterday, W.R.; Huynh, L.; U’Ren, J.M.; Dukerich, M.; Zanecki, S.R.; Kenefic, L.J.; Beaudry, J.; et al. Bacillus anthracis in China and its relationship to worldwide lineages. BMC Microbiol. 2009, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Timofeev, V.; Bahtejeva, I.; Mironova, R.; Titareva, G.; Lev, I.; Christiany, D.; Borzilov, A.; Bogun, A.; Vergnaud, G. Insights from Bacillus anthracis strains isolated from permafrost in the tundra zone of Russia. PLoS ONE 2019, 4, e0209140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, T.; Busch, J.D.; Ravel, J.; Read, T.D.; Rhoton, S.D.; U’Ren, J.M.; Simonson, T.S.; Kachur, S.M.; Leadem, R.R.; Cardon, M.L.; et al. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2004, 101, 13536–13541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, M.; Buyuk, F.; Baillie, L.; Wölfel, R.; Kotorashvili, A.; Rehn, A.; Antwerpen, M.; Grass, G. The identification of novel single nucleotide polymorphisms to assist in mapping the spread of Bacillus anthracis across the Southern Caucasus. Sci. Rep. 2018, 8, 11254. [Google Scholar] [CrossRef] [Green Version]
- Derzelle, S.; Aguilar-Bultet, L.; Frey, J. Comparative genomics of Bacillus anthracis from the wool industry highlights polymorphisms of lineage A.Br.Vollum. Infect. Genet. Evol. 2016, 46, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Wiedmann, M.; Bruce, J.L.; Keating, C.; Johnson, A.E.; McDonough, P.L.; Batt, C.A. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect. Immun. 1997, 65, 2707–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakov, A.V.; Mastriani, E.; Liu, S.L.; Schifferli, D.M. Association of Salmonella virulence factor alleles with intestinal and invasive serovars. BMC Genom. 2019, 20, 429. [Google Scholar] [CrossRef]
- Springman, A.C.; Lacher, D.W.; Wu, G.; Milton, N.; Whittam, T.S.; Davies, H.D.; Manning, S.D. Selection, recombination, and virulence gene diversity among group B streptococcal genotypes. J. Bacteriol. 2009, 191, 5419–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, A.P.; Dentovskaya, S.V.; Panfertsev, E.A.; Svetoch, T.E.; Kopylov, P.K.; Segelke, B.W.; Zemla, A.; Telepnev, M.V.; Motin, V.L. Amino acid and structural variability of Yersinia pestis LcrV protein. Infect. Genet. Evol. 2010, 10, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Dentovskaya, S.V.; Platonov, M.E.; Svetoch, T.E.; Kopylov, P.K.; Kombarova, T.I.; Ivanov, S.A.; Shaikhutdinova, R.Z.; Kolombet, L.V.; Chauhan, S.; Ablamunits, V.G.; et al. Two Isoforms of Yersinia pestis Plasminogen Activator Pla: Intraspecies Distribution, Intrinsic Disorder Propensity, and Contribution to Virulence. PLoS ONE 2016, 11, e0168089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopylov, P.K.; Platonov, M.E.; Ablamunits, V.G.; Kombarova, T.I.; Ivanov, S.A.; Kadnikova, L.A.; Somov, A.N.; Dentovskaya, S.V.; Uversky, V.N.; Anisimov, A.P. Yersinia pestis Caf1 Protein: Effect of Sequence Polymorphism on Intrinsic Disorder Propensity, Serological Cross-Reactivity and Cross-Protectivity of Isoforms. PLoS ONE 2016, 11, e0162308. [Google Scholar] [CrossRef]
- Price, L.B.; Hugh-Jones, M.; Jackson, P.J.; Keim, P. Genetic diversity in the protective antigen gene of Bacillus anthracis. J. Bacteriol. 1999, 181, 2358–2362. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carriço, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef]
- Pannifer, A.D.; Wong, T.Y.; Schwarzenbacher, R.; Renatus, M.; Petosa, C.; Bienkowska, J.; Lacyk, D.B.; Collier, R.J.; Park, S.; Leppla, S.H.; et al. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Drum, C.L.; Yan, S.-Z.; Bard, J.; Shen, Y.-Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.-J. Structural Basis for the Activation of Anthrax Adenylyl Cyclase Exotoxin by Calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carrico, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Okinaka, R.T.; Henrie, M.; Hill, K.K.; Lowery, K.S.; Van Ert, M.; Pearson, T.; Schupp, J.; Kenefic, L.; Beaudry, J.; Hofstadler, S.A.; et al. Single nucleotide polymorphism typing of Bacillus anthracis from Sverdlovsk tissue. Emerg. Infect. Dis. 2008, 14, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Eremenko, E.I.; Ryazanova, A.G.; Pisarenko, S.V.; Aksenova Yu, L.; Semenova, O.V.; Koteneva, E.A.; Tsygankova, O.I.; Kovalev, D.A.; Golovinskaya, T.M.; Chmerenko, D.K.; et al. Comparative Analysis of Genetic Typing Methods for Bacillus anthracis. Russ. J. Genet. 2019, 55, 40–51. [Google Scholar] [CrossRef]
- Pisarenko, S.V.; Eremenko, E.I.; Ryazanova, A.G.; Kovalev, D.A.; Buravtseva, N.P.; Aksenova, L.Y.; Dugarzhapova, Z.F.; Evchenko, A.Y.; Kravets, E.V.; Semenova, O.V.; et al. Phylogenetic analysis of Bacillus anthracis strains from Western Siberia reveals a new genetic cluster in the global population of the species. BMC Genom. 2019, 20, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Qi, X.; Zhu, L.; Wang, D.; Gao, Z.; Deng, H.; Wu, W.; Hu, T.; Chen, C.; Chen, W.; et al. Genome sequence of Bacillus anthracis attenuated vaccine strain A16R used for human in China. J. Biotechnol. 2015, 210, 15–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne, M. The use of anthrax vaccines prepared from avirulent (uncapsulated) variants of Bacillus anthracis. Onderstepoort J. Vet. Sci. Anim. Ind. 1939, 13, 307–312. [Google Scholar]
- Cohen-Gihon, I.; Israeli, O.; Beth-Din, A.; Levy, H.; Cohen, O.; Shafferman, A.; Zvi, A.; Chitlaru, T. Whole-Genome Sequencing of the Nonproteolytic Bacillus anthracis V770-NP1-R Strain Reveals Multiple Mutations in Peptidase Loci. Genome Announc. 2014, 2, e00075-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellows, P.F.; Linscott, M.K.; Ivins, B.E.; Pitt, M.L.; Rossi, C.A.; Gibbs, P.H.; Friedlander, A.M. Efficacy of a human anthrax vaccine in guinea pigs, rabbits, and rhesus macaques against challenge by Bacillus anthracis isolates of diverse geographical origin. Vaccine 2001, 19, 3241–3247. [Google Scholar] [CrossRef]
- Turnbull, P.C. Anthrax vaccines: Past, present and future. Vaccine 1991, 9, 533–539. [Google Scholar] [CrossRef]
- Lindeque, P.M.; Turnbull, P.C. Ecology and epidemiology of anthrax in the Etosha National Park, Namibia. Onderstepoort J. Vet. Sci. Anim. Ind. 1994, 61, 71–83. [Google Scholar]
- De Vos, V. The ecology of anthrax in the Kruger National Park, South Africa. Salisb. Med Bull. 1990, 68, 19–23. [Google Scholar]
- Beyer, W.; Bellan, S.; Eberle, G.; Ganz, H.H.; Getz, W.M.; Haumacher, R.; Hilss, K.A.; Kilian, W.; Lazak, J.; Turner, W.C.; et al. Distribution and molecular evolution of Bacillus anthracis genotypes in Namibia. PLoS Negl. Trop. Dis. 2012, 6, e1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnaud, G.; Girault, G.; Thierry, S.; Pourcel, C.; Madani, N.; Blouin, Y. Comparison of French and Worldwide Bacillus anthracis Strains Favors a Recent, Post-Columbian Origin of the Predominant North-American Clade. PLoS ONE 2016, 11, e0146216. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).