
Analysis of In Vivo Transcriptome of Intracellular Bacterial Pathogen Salmonella enterica serovar Typhmurium Isolated from Mouse Spleen




Abstract
:1. Introduction
2. Results
2.1. Gene Expression Profiles
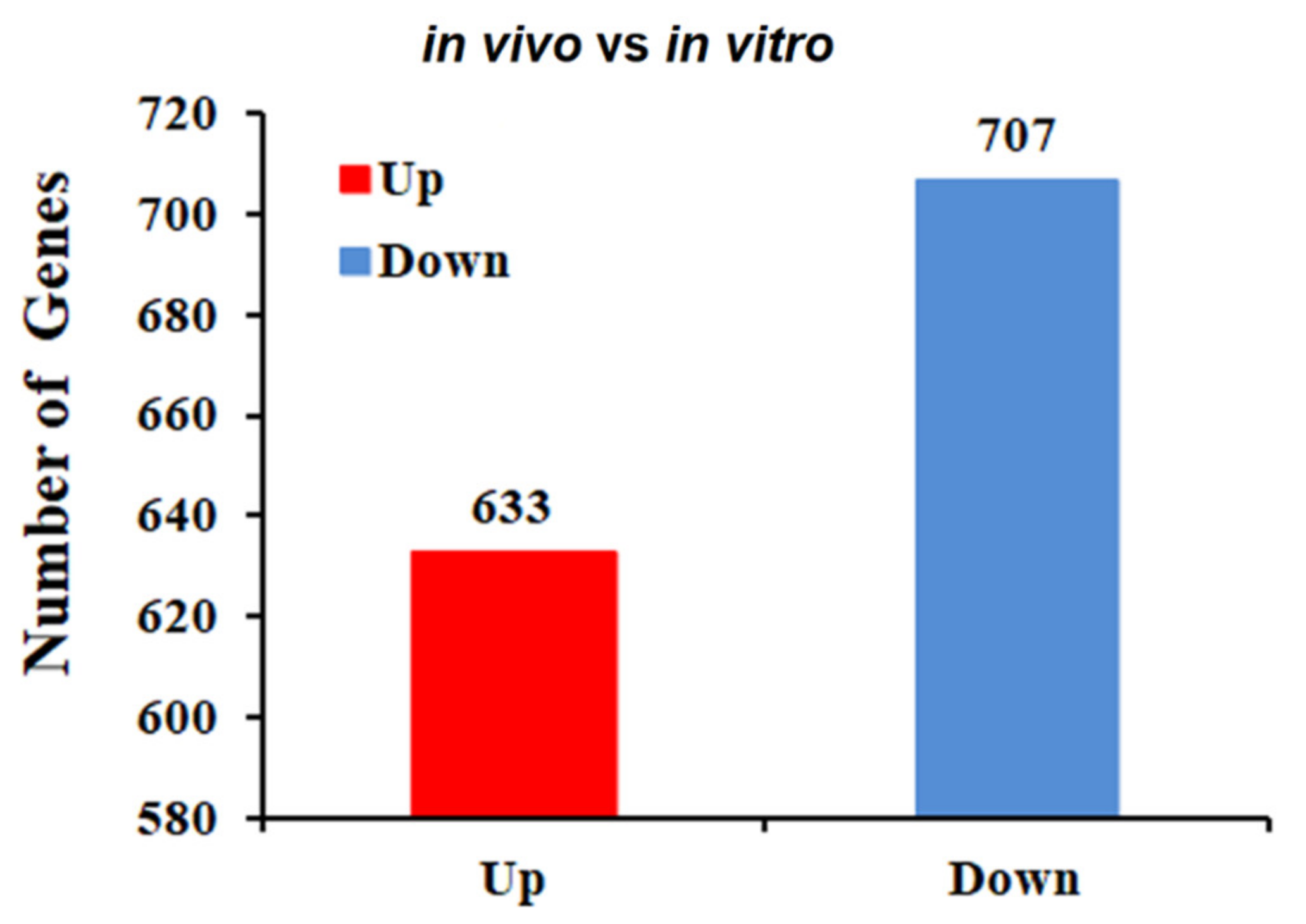
2.2. Identification of DEGs
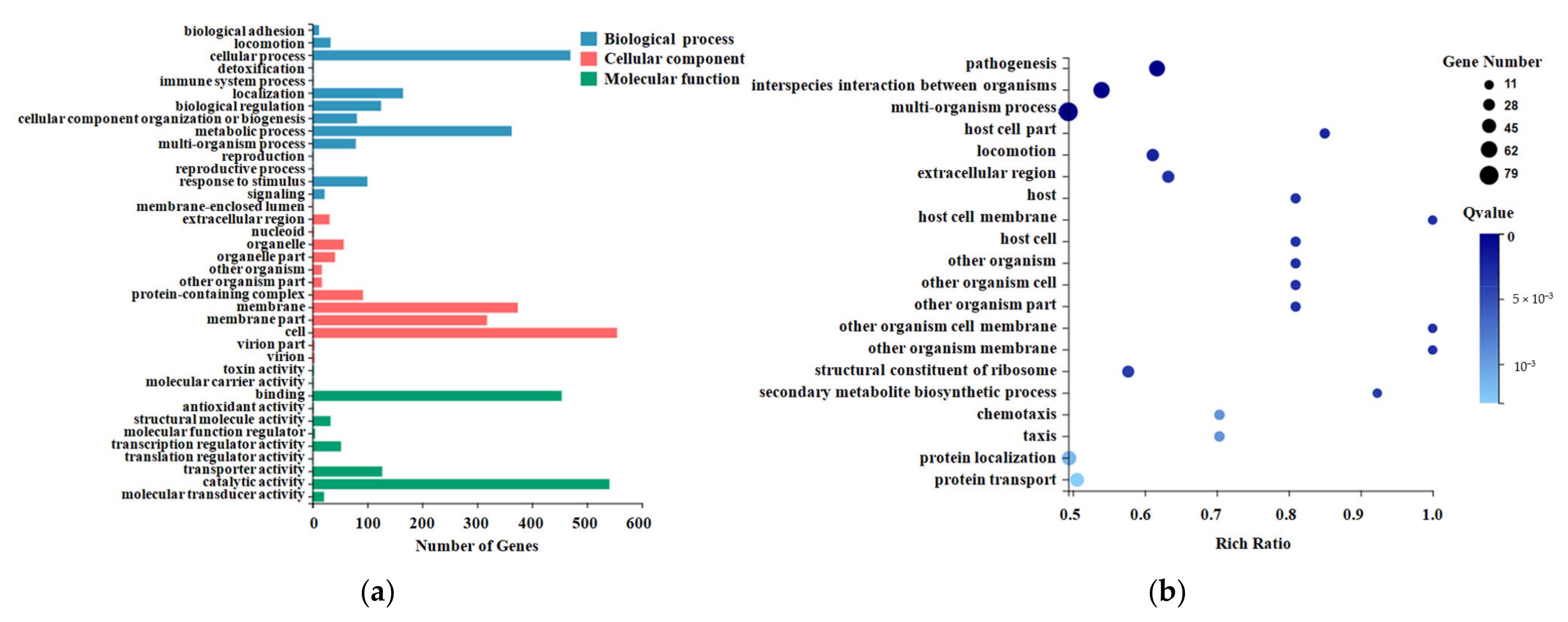
2.3. Gene Ontology (GO) Enrichment Analysis for DEGs
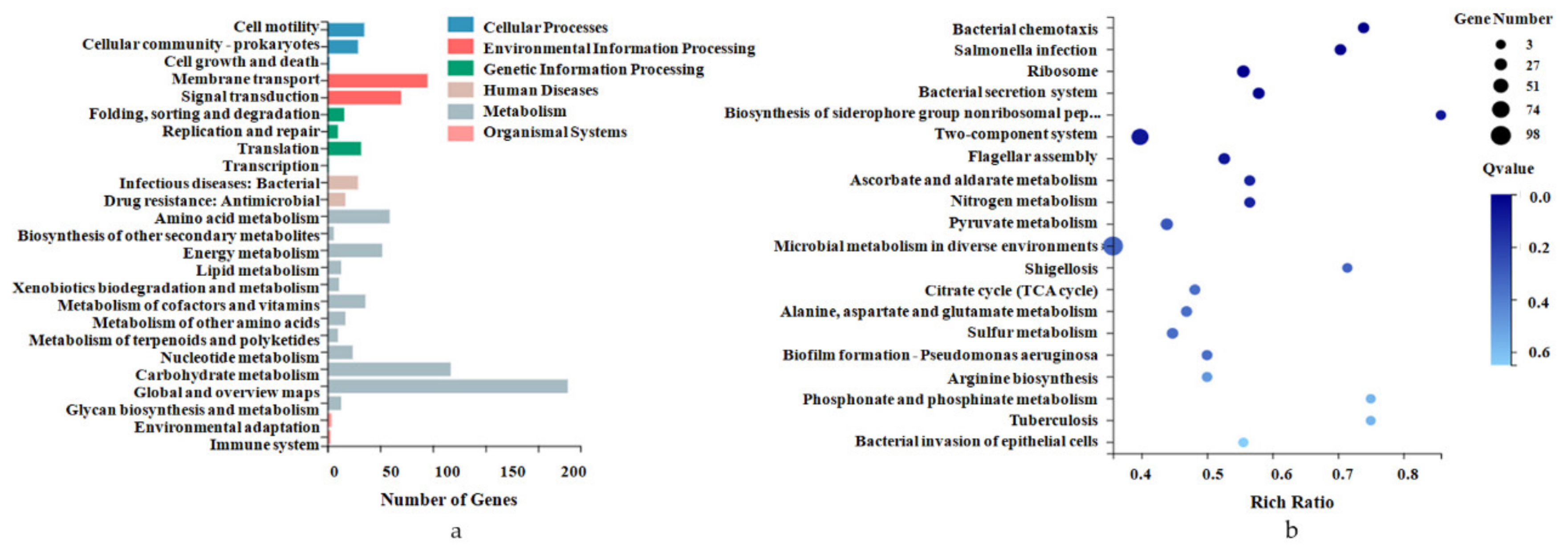
2.4. Pathway Enrichment Analysis for DEGs
2.5. Validation of the RNA-seq Data by qPCR
2.6. Screening of CKGs for Survival and Proliferation In Vivo
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Animals
4.2. BALB/c Mice Infection and RNA Extraction
4.3. Library Construction and RNA Sequencing
4.4. Identification of Differentially Expressed Genes (DEGs)
4.5. Gene Ontology (GO) and Pathway Enrichment Analysis for DEGs
4.6. Quantitative PCR Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Egal-Mor, O.; Boyle, E.C.; Grassl, G.A. Same species, different diseases: How and why typhoidal and non-typhoidal Salmonella enterica serovars differ. Front. Microbiol. 2014, 5, 391. [Google Scholar] [CrossRef] [Green Version]
- Ansari, N.; Yazdian-Robati, R.; Shahdordizadeh, M.; Wang, Z.; Ghazvini, K. Aptasensors for quantitative detection of Salmonella Typhimurium. Anal. Biochem. 2017, 533, 18–25. [Google Scholar] [CrossRef]
- Watson, K.G.; Holden, D.W. Dynamics of growth and dissemination of Salmonella in vivo. Cell. Microbiol. 2010, 12, 1389–1397. [Google Scholar] [CrossRef]
- Braukmann, M.; Methner, U.; Berndt, A. Immune Reaction and Survivability of Salmonella Typhimurium and Salmonella Infantis after Infection of Primary Avian Macrophages. PLoS ONE 2015, 10, e0122540. [Google Scholar] [CrossRef] [Green Version]
- Ricchi, M.; Mazzarelli, A.; Piscini, A.; Di Caro, A.; Cannas, A.; Leo, S.; Russo, S.; Arrigoni, N. Exploring MALDI-TOF MS approach for a rapid identification of Mycobacterium avium ssp. paratuberculosis field isolates. J. Appl. Microbiol. 2017, 122, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chen, Y.; Xie, X.; Xia, J.; Li, Q.; Geng, S.; Jiao, X. Influence of Salmonella enterica serovar Pullorum pathogenicity island 2 on type III secretion system effector gene expression in chicken macrophage HD11 cells. Avian Pathol. 2016, 46, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, T.; Fieselmann, A.; Fischer, E.; Popp, J.; Hensel, M.; Noster, J. Salmonella-how a metabolic generalist adopts an intracellular lifestyle during infection. Fron. Cell. Infect. Microbiol. 2015, 4, 191. [Google Scholar]
- Shen, S.; Fang, F.C. Integrated stress responses in Salmonella. Int. J. Food Microbiol. 2012, 152, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Hu, H.; Chavda, K.D.; Zhao, S.; Liu, R.; Liang, H.; Zhang, W.; Wang, X.; Jacobs, M.R.; Bonomo, R.A.; et al. Complete Sequence of a KPC-Producing IncN Multidrug-Resistant Plasmid from an Epidemic Escherichia coli Sequence Type 131 Strain in China. Antimicrob. Agents Chemother. 2014, 58, 2422–2425. [Google Scholar] [CrossRef] [Green Version]
- Ellis, T.C.; Jain, S.; Linowski, A.J.; Rike, K.; Bestor, A.; Rosa, P.A.; Halpern, M.; Kurhanewicz, S.; Jewett, M.W. In vivo expression technology identifies a novel virulence factor critical for Borrelia burgdorferi persistence in mice. PLoS Pathog. 2013, 9, e1003567. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, J.; Li, Q.; Shang, Y.; Ren, F.; Jiao, Y.; Liu, Z.; Pan, Z.; Jiao, X.-A. Use of in vivo-induced antigen technology to identify in vivo-expressed genes of Campylobacter jejuni during human infection. J. Microbiol. Biotechnol. 2014, 24, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Casselli, T.; Bankhead, T. Use of in vivo Expression Technology for the Identification of Putative Host Adaptation Factors of the Lyme Disease Spirochete. J. Mol. Microbiol. Biotechnol. 2015, 25, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Ron, M.; Gorelick-Ashkenazi, A.; Levisohn, S.; Nir-Paz, R.; Geary, S.J.; Tulman, E.; Lysnyansky, I.; Yogev, D. Mycoplasma gallisepticum in vivo induced antigens expressed during infection in chickens. Vet. Microbiol. 2015, 175, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Galán, J.E.; Wolf-Watz, H. Protein delivery into eukaryotic cells by type III secretion machines. Nat. Cell Biol. 2006, 444, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Soncini, F.C.; Solomon, F.; Groisman, E.A. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. USA 1996, 93, 7800–7804. [Google Scholar] [CrossRef] [Green Version]
- Lara-Tejero, M.; Galán, J.E. Salmonella enterica Serovar Typhimurium Pathogenicity Island 1-Encoded Type III Secretion System Translocases Mediate Intimate Attachment to Nonphagocytic Cells. Infect. Immun. 2009, 77, 2635–2642. [Google Scholar] [CrossRef] [Green Version]
- Giacomodonato, M.N.; Llana, M.N.; Castaneda, M.D.R.A.; Buzzola, F.R.; Sarnacki, S.H.; Cerquetti, M.C. AvrA effector protein of Salmonella enterica serovar Enteritidis is expressed and translocated in mesenteric lymph nodes at late stages of infection in mice. Microbiology 2014, 160, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Cong, Y.; Li, S.; Rao, X.; Wang, G.; Hu, F. Identification of in vivo induced protein antigens of Salmonella enterica serovar Typhi during human infection. Sci. China Ser. C Life Sci. 2009, 52, 942–948. [Google Scholar] [CrossRef]
- Alam, M.M.; Tsai, L.L.; Rollins, S.M.; Sheikh, A.; Khanam, F.; Bufano, M.K.; Yu, Y.; Wu-Freeman, Y.; Kalsy, A.; Sultana, T.; et al. Identification of in vivo-induced bacterial proteins during human infection with Salmonella enterica serotype Paratyphi A. Clin. Vaccine Immunol. 2013, 20, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Liu, Z.; Lin, Z.; Barrow, P.; Pan, Z.; Li, Q.; Jiao, X. Identification of in vivo-induced genes during infection of chickens with Salmonella enterica serovar Enteritidis. Res. Vet. Sci. 2015, 100, 1–7. [Google Scholar] [CrossRef]
- Geng, S.; Wang, Y.; Xue, Y.; Wang, H.; Cai, Y.; Zhang, J.; Barrow, P.; Pan, Z.; Jiao, X. The SseL protein inhibits the intracellular NF-kappaB pathway to enhance the virulence of Salmonella Pullorum in a chicken model. Microb. Pathog. 2019, 129, 1–6. [Google Scholar] [CrossRef]
- Mahan, M.J.; Slauch, J.M.; Mekalanos, J.J. Selection of bacterial virulence genes that are specifically induced in host tissues. Science 1993, 259, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Cacaci, M.; Sauvageot, N.; Sanguinetti, M.; Rattei, T.; Eder, T.; Giard, J.-C.; Kalinowski, J.; Hain, T.; Hartke, A. The Intraperitoneal Transcriptome of the Opportunistic Pathogen Enterococcus faecalis in Mice. PLoS ONE 2015, 10, e0126143. [Google Scholar] [CrossRef]
- Eriksson, S.; Lucchini, S.; Thompson, A.; Rhen, M.; Hinton, J. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 2003, 47, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Cummings, L.A.; Wilkerson, W.D.; Bergsbaken, T.; Cookson, B.T. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol. Microbiol. 2006, 61, 795–809. [Google Scholar] [CrossRef]
- Browne, S.H.; Lesnick, M.L.; Guiney, D.G. Genetic requirements for salmonella-induced cytopathology in human monocyte-derived macrophages. Infect. Immun. 2002, 70, 7126–7135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.-J.; Grabe, G.J.; Liu, M.; Mota, L.J.; Holden, D.W. SsaV Interacts with SsaL to Control the Translocon-to-Effector Switch in the Salmonella SPI-2 Type Three Secretion System. mBio 2018, 9, e01149-18. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.-J.; Liu, M.; Holden, D.W. SsaM and SpiC interact and regulate secretion of Salmonella Pathogenicity Island 2 type III secretion system effectors and translocators. Mol. Microbiol. 2004, 54, 604–619. [Google Scholar] [CrossRef]
- Yoshida, Y.; Miki, T.; Ono, S.; Haneda, T.; Ito, M.; Okada, N. Functional Characterization of the Type III Secretion ATPase SsaN Encoded by Salmonella Pathogenicity Island 2. PLoS ONE 2014, 9, e94347. [Google Scholar] [CrossRef]
- Ge, S.; He, Q.; Granfors, K. HLA-B27 Modulates Intracellular Growth of Salmonella Pathogenicity Island 2 Mutants and Production of Cytokines in Infected Monocytic U937 Cells. PLoS ONE 2012, 7, e34093. [Google Scholar] [CrossRef]
- Cooper, C.A.; Mulder, D.T.; Allison, S.E.; Pilar, A.V.C.; Coombes, B.K. The SseC translocon component in Salmonella enterica serovar Typhimurium is chaperoned by SscA. BMC Microbiol. 2013, 13, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, R.L.; Gonias, L.A.; Stein, M.A. Aggregation of host endosomes by Salmonella requires SPI2 translocation of SseFG and involves SpvR and the fms-aroE intragenic region. Mol. Microbiol. 2000, 37, 1417–1435. [Google Scholar] [CrossRef]
- Dai, S.; Zhou, D. Secretion and Function of Salmonella SPI-2 Effector SseF Require Its Chaperone, SscB. J. Bacteriol. 2004, 186, 5078–5086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurawski, D.; Stein, M.A. SseA acts as the chaperone for the SseB component of the Salmonella Pathogenicity Island 2 translocon. Mol. Microbiol. 2003, 47, 1341–1351. [Google Scholar] [CrossRef]
- Hensel, M.; Shea, J.E.; Waterman, S.R.; Mundy, R.; Nikolaus, T.; Banks, G.; Vazques-Torres, A.; Gleeson, C.; Fang, F.C.; Holden, D.W. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 1998, 30, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Peng, D.; Jiao, X.; Zhang, X.; Geng, S.; Liu, X. Roles of the spiA gene from Salmonella enteritidis in biofilm formation and virulence. Microbiology 2011, 157, 1798–1805. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.A.; Rappl, C.; Kuhle, V.; Hensel, M.; Miller, S.I. SpiC Is Required for Translocation of Salmonella Pathogenicity Island 2 Effectors and Secretion of Translocon Proteins SseB and SseC. J. Bacteriol. 2002, 184, 4971–4980. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.-J.; Ruiz-Albert, J.; Unsworth, K.E.; Garvis, S.; Liu, M.; Holden, D.W. SpiC is required for secretion of Salmonella Pathogenicity Island 2 type III secretion system proteins. Cell. Microbiol. 2002, 4, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, P.P.; Devegowda, D.; Ruwandeepika, H.A.D.; Karunasagar, I.; Karunasagar, I. Presence of Salmonella pathogenicity island 2 genes in seafood-associated Salmonella serovars and the role of the sseC gene in survival of Salmonella enterica serovar Weltevreden in epithelial cells. Microbiology 2011, 157, 160–168. [Google Scholar] [CrossRef]
- Maserati, A.; Fink, R.C.; Lourenco, A.; Julius, M.; Diez-Gonzalez, F. General response of Salmonella enterica serovar Typhimurium to desiccation: A new role for the virulence factors sopD and sseD in survival. PLoS ONE 2017, 12, e0187692. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.-Z.; Jiang, A.-J.; Mao, A.-W.; Feng, Y.; Wang, W.; Li, J.; Zhang, X.; Xing, K.; Peng, X. The Salmonella effectors SseF and SseG inhibit Rab1A-mediated autophagy to facilitate intracellular bacterial survival and replication. J. Biol. Chem. 2018, 293, 9662–9673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzzau, S.; Brown, D.; Wallis, T.; Rubino, S.; Leori, G.; Bernard, S.; Casadesús, J.; Platt, D.J.; Olsen, J.E. Host adapted serotypes of Salmonella enterica. Epidemiol. Infect. 2000, 125, 229–255. [Google Scholar] [CrossRef]
- Shi, L.; Ansong, C.; Smallwood, H.; Rommereim, L.; McDermott, J.; Brewer, H.M.; Norbeck, A.D.; Taylor, R.C.; Gustin, J.K.; Heffron, F.; et al. Proteome of Salmonella Enterica Serotype Typhimurium Grown in a Low Mg2+/pH Medium. J. Proteom. Bioinform. 2009, 2, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Goto, R.; Miki, T.; Nakamura, N.; Fujimoto, M.; Okada, N. Salmonella Typhimurium PagP- and UgtL-dependent resistance to antimicrobial peptides contributes to the gut colonization. PLoS ONE 2017, 12, e0190095. [Google Scholar]
- Choi, J.; Groisman, E.A. Activation of master virulence regulator PhoP in acidic pH requires the Salmonella-specific protein UgtL. Sci. Signal. 2017, 10, 6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouslim, C.; Groisman, E.A. Control of the Salmonella ugd gene by three two-component regulatory systems. Mol. Microbiol. 2003, 47, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Quezada, C.M.; Hicks, S.W.; Galán, J.E.; Stebbins, C.E. A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2009, 106, 4864–4869. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.S.; Gunn, J.S.; Morona, R. The Sahonella typhi melittin resistance gene pgaB affects intracellular growth in PMA-differentiated U937 cells, polymyxin B resistance and lipopolysaccharide. Microbiology 1999, 145, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Detweiler, C.S.; Monack, D.M.; Brodsky, I.E.; Mathew, H.; Falkow, S. virK, somA and rcsC are important for systemic Salmonella enterica serovar Typhimurium infection and cationic peptide resistance. Mol. Microbiol. 2003, 48, 385–400. [Google Scholar] [CrossRef]
- Brodsky, I.E.; Ghori, N.; Falkow, S.; Monack, D. Mig-14 is an inner membrane-associated protein that promotes Salmonella typhimurium resistance to CRAMP, survival within activated macrophages and persistent infection. Mol. Microbiol. 2004, 55, 954–972. [Google Scholar] [CrossRef]
- Gall, A.R.; Hegarty, A.E.; Datsenko, K.A.; Westerman, R.P.; SanMiguel, P.; Csonka, L.N. High-level, constitutive expression of the mgtC gene confers increased thermotolerance on Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2018, 109, 327–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Nam, D.; Kweon, D.-H.; Shin, D. ATP reduction by MgtC and Mg2+ homeostasis by MgtA and MgtB enables Salmonella to accumulate RpoS upon low cytoplasmic Mg2+ stress. Mol. Microbiol. 2018, 110, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Chekabab, S.M.; Yu, H.; Yin, X.; Diarra, M.S.; Yang, C.; Gong, J. Growth and Virulence of Salmonella Typhimurium Mutants Deficient in Iron Uptake. ACS Omega 2019, 4, 13218–13230. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Du, W.; Wang, M.; Li, Y.; Su, S.; Wu, T.; Kang, Y.; Shan, X.; Shi, Q.; Zhu, G. Contribution of the colicin receptor CirA to biofilm formation, antibotic resistance, and pathogenicity of Salmonella Enteritidis. J. Basic Microbiol. 2019, 60, 72–81. [Google Scholar] [CrossRef]
- Knodler, L.A.; Vallance, B.A.; Hensel, M.; Jäckel, D.; Finlay, B.B.; Steele-Mortimer, O. Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol. Microbiol. 2004, 49, 685–704. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, N.; Henry, T.; De Chastellier, C.; Zhao, W.; Guilhon, A.-A.; Gorvel, J.-P.; Méresse, S. The Virulence Protein SopD2 Regulates Membrane Dynamics of Salmonella-Containing Vacuoles. PLoS Pathog. 2010, 6, e1001002. [Google Scholar] [CrossRef]
- Knodler, L.A.; Steele-Mortimer, O. The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol. Biol. Cell. 2005, 16, 4108–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III Secretion Systems and Disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef] [Green Version]
- Schleker, S.; Sun, J.; Raghavan, B.; Srnec, M.; Müller, N.; Koepfinger, M.; Murthy, L.; Zhao, Z.; Klein-Seetharaman, J. The current Salmonella -host interactome. Proteom. Clin. Appl. 2012, 6, 117–133. [Google Scholar] [CrossRef]
- Pinaud, L.; Sansonetti, P.J.; Phalipon, A. Host Cell Targeting by Enteropathogenic Bacteria T3SS Effectors. Trends Microbiol. 2018, 26, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Jennings, E.; Thurston, T.L.; Holden, D.W. Salmonella SPI-2 Type III Secretion System Effectors: Molecular Mechanisms and Physiological Consequences. Cell Host Microbe 2017, 22, 217–231. [Google Scholar] [CrossRef]
- Shao, F. Mechanisms of Interactions between Bacterial Pathogen and Host Innate Immune Defense System. Available online: https://en.cnki.com.cn/Article_en/CJFDTotal-ZGJB201201005.htm (accessed on 29 June 2021).
- Jones, R.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 2008, 3, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.R.; Jones, B.D. Salmonella pathogenicity island 2-encoded proteins SseC and SseD are essential for virulence and are substrates of the type III secretion system. Infect. Immun. 2001, 69, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Tong, L. Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci. 2013, 70, 863–891. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Li, D.-F.; Wang, D.-C.; Fleming, J.; Zhang, H.; Zhou, Y.; Zhou, L.; Zhou, J.; Chen, T.; Chen, G.; et al. Structure and function of Mycobacterium smegmatis 7-keto-8-aminopelargonic acid (KAPA) synthase. Int. J. Biochem. Cell Biol. 2015, 58, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Goldberg, M.D.; Carroll, R.K.; Danino, V.; Hinton, J.; Dorman, C.J. A global role for Fis in the transcriptional control of metabolism and type III secretion in Salmonella enterica serovar Typhimurium. Microbiology 2004, 150, 2037–2053. [Google Scholar] [CrossRef]
- Kubota, T.; Izumi, Y. Detection and Characterization of a Thermophilic Biotin Biosynthetic Enzyme, 7-Keto-8-aminopelargonic Acid Synthase, from Various Thermophiles. Biosci. Biotechnol. Biochem. 2012, 76, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Li, D.; Fleming, J.; Hong, Y.; Chen, T.; Zhou, L.; Bi, L.; Wang, D.; Zhang, X.; Chen, G. Purification and X-ray crystallographic analysis of 7-keto-8-aminopelargonic acid (KAPA) synthase from Mycobacterium smegmatis. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1372–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucciarelli, M.G.; Portillo, F.G.-D. Salmonella Intracellular Lifestyles and Their Impact on Host-to-Host Transmission. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Rowley, G.; Hensen, D.; Felgate, H.; Arkenberg, A.; Appia-Ayme, C.; Prior, K.; Harrington, C.; Field, S.J.; Butt, J.N.; Baggs, L.; et al. Resolving the contributions of the membrane-bound and periplasmic nitrate reductase systems to nitric oxide and nitrous oxide production in Salmonella enterica serovar Typhimurium. Biochem. J. 2011, 441, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Helaine, S.; Holden, D.W. Heterogeneity of intracellular replication of bacterial pathogens. Curr. Opin. Microbiol. 2013, 16, 184–191. [Google Scholar] [CrossRef]
- Aguilar-Barajas, E.; Pérez, C.D.; Ramirez-Diaz, M.I.; Riveros-Rosas, H.; Cervantes, C. Bacterial transport of sulfate, molybdate, and related oxyanions. BioMetals 2011, 24, 687–707. [Google Scholar] [CrossRef]
- Zolotarev, A.S.; Unnikrishnan, M.; Shmukler, B.E.; Clark, J.S.; Vandorpe, D.H.; Grigorieff, N.; Rubin, E.; Alper, S.L. Increased sulfate uptake by E. coli overexpressing the SLC26-related SulP protein Rv1739c from Mycobacterium tuberculosis. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2008, 149, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Senaratne, R.H.; De Silva, A.D.; Williams, S.J.; Mougous, J.D.; Reader, J.R.; Zhang, T.; Chan, S.; Sidders, B.; Lee, D.H.; Chan, J.; et al. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol. Microbiol. 2006, 59, 1744–1753. [Google Scholar] [CrossRef]
- Richardson, A.R.; Payne, E.C.; Younger, N.; Karlinsey, J.E.; Thomas, V.C.; Becker, L.A.; Navarre, W.W.; Castor, M.E.; Libby, S.J.; Fang, F.C. Multiple Targets of Nitric Oxide in the Tricarboxylic Acid Cycle of Salmonella enterica Serovar Typhimurium. Cell Host Microbe 2011, 10, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.; McDermott, J.E.; Porwollik, S.; McClelland, M.; Heffron, F. Coordinated Regulation of Virulence during Systemic Infection of Salmonella enterica Serovar Typhimurium. PLoS Pathog. 2009, 5, e1000306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikumar, S.; Kröger, C.; Hébrard, M.; Colgan, A.; Owen, S.; Sivasankaran, S.K.; Cameron, A.; Hokamp, K.; Hinton, J.C.D. RNA-seq Brings New Insights to the Intra-Macrophage Transcriptome of Salmonella Typhimurium. PLoS Pathog. 2015, 11, e1005262. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.Y.; Hall, J.M.; Nowak, E.S.; Boehm, D.T.; Gonyar, L.A.; Hewlett, E.L.; Eby, J.C.; Barbier, M.; Damron, F.H. Analysis of the In Vivo Transcriptome of Bordetella pertussis during Infection of Mice. mSphere 2019, 4, 154–219. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzov, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, M.P.D.K.G.M.P.S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D. The Gene Ontology and the Meaning of Biological Function. Methods Mol. Biol. 2017, 1446, 15–24. [Google Scholar]
- Minoru, K.; Furumich, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, 353–361. [Google Scholar]
- Garrido-Franco, M. Pyridoxine 5prime-phosphate synthase: De novo synthesis of vitamin B6 and beyond. Biochim. Biophys. Acta 2003, 1647, 92–97. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(–delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Description | Number | Gene Symbol |
|---|---|---|
| T3SS-associated protein | 17 | ssa (D, H, I, J [26], L [27], M [28], N [29], P, S [30], T, U, and V [26,27]), sscA [30,31], sscB [32,33], sseA [34,35]), yscR, spiA [36]; |
| Pathogenicity-island-2-secreted effector protein | 7 | spiC [37,38], sseB [35], sseC [39], sseD [40], sseE [32,35], sseF [35,41], STM1410; |
| Hypothetical protein | 7 | steC, SseL [21,42], STM2138, yncJ, ssaK, pagO, pagC |
| Enzyme | 12 | bioF, bioB [43], bioA, bioD, ugtl [44,45], ugd [46], STM1952, cysH, sspH2 [47], entC, Fe-S, pqaB [48]; |
| Antimicrobial resistance protein | 3 | virK [49], mig-14 [50], pagP [44]; |
| Transporter | 5 | mgtC [51], mgtB [52], rbsA, cysA, fruB; |
| Receptor protein | 3 | fepA [53], cirA [54], iroN [53] |
| Cold shock protein | 1 | cspH; |
| Effector protein | 3 | sifB, pipB2 [55,56], pipB [57]; |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, N.; Song, Y.; Liu, C.; Liu, M.; Yu, L.; Wang, F. Analysis of In Vivo Transcriptome of Intracellular Bacterial Pathogen Salmonella enterica serovar Typhmurium Isolated from Mouse Spleen. Pathogens 2021, 10, 823. https://doi.org/10.3390/pathogens10070823
Sun N, Song Y, Liu C, Liu M, Yu L, Wang F. Analysis of In Vivo Transcriptome of Intracellular Bacterial Pathogen Salmonella enterica serovar Typhmurium Isolated from Mouse Spleen. Pathogens. 2021; 10(7):823. https://doi.org/10.3390/pathogens10070823
Chicago/Turabian StyleSun, Na, Yanying Song, Cong Liu, Mengda Liu, Lanping Yu, and Fangkun Wang. 2021. "Analysis of In Vivo Transcriptome of Intracellular Bacterial Pathogen Salmonella enterica serovar Typhmurium Isolated from Mouse Spleen" Pathogens 10, no. 7: 823. https://doi.org/10.3390/pathogens10070823
APA StyleSun, N., Song, Y., Liu, C., Liu, M., Yu, L., & Wang, F. (2021). Analysis of In Vivo Transcriptome of Intracellular Bacterial Pathogen Salmonella enterica serovar Typhmurium Isolated from Mouse Spleen. Pathogens, 10(7), 823. https://doi.org/10.3390/pathogens10070823