
The History of Anti-Trypanosome Vaccine Development Shows That Highly Immunogenic and Exposed Pathogen-Derived Antigens Are Not Necessarily Good Target Candidates: Enolase and ISG75 as Examples

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Parasites Infections
2.2. Recombinant Production and Purification of TbENO
2.3. Recombinant Production and Purification of TbgISG75
2.4. Circular Dichroism (CD) Spectroscopy
2.5. Small-Angle X-ray Scattering (SAXS) Data Collection and Processing
2.6. Isothermal Titration Calorimetry (ITC)
2.7. Vaccine Procedure and In Vivo T. b. brucei Challenge
2.8. Quantification of Anti-TbENO and Anti-TbgISG75 Antibody Titers by ELISA
2.9. Cell Preparation and Flow Cytometry Analysis
2.10. Flow Cytometry Detection Reagents
2.11. Statistical Analysis
3. Results
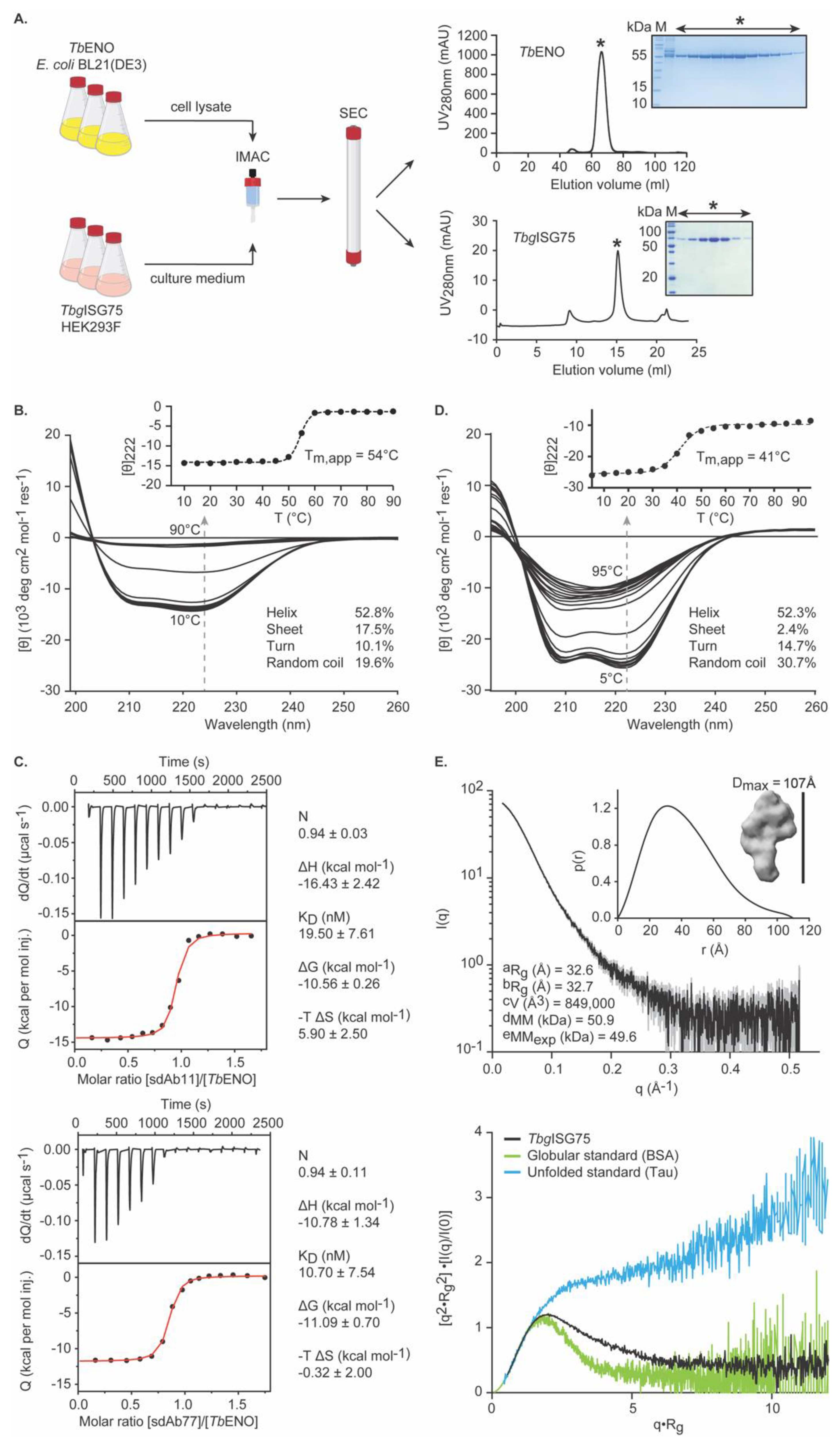
3.1. Recombinant TbENO and TbgISG75 Are Correctly Folded and Biologically Relevant
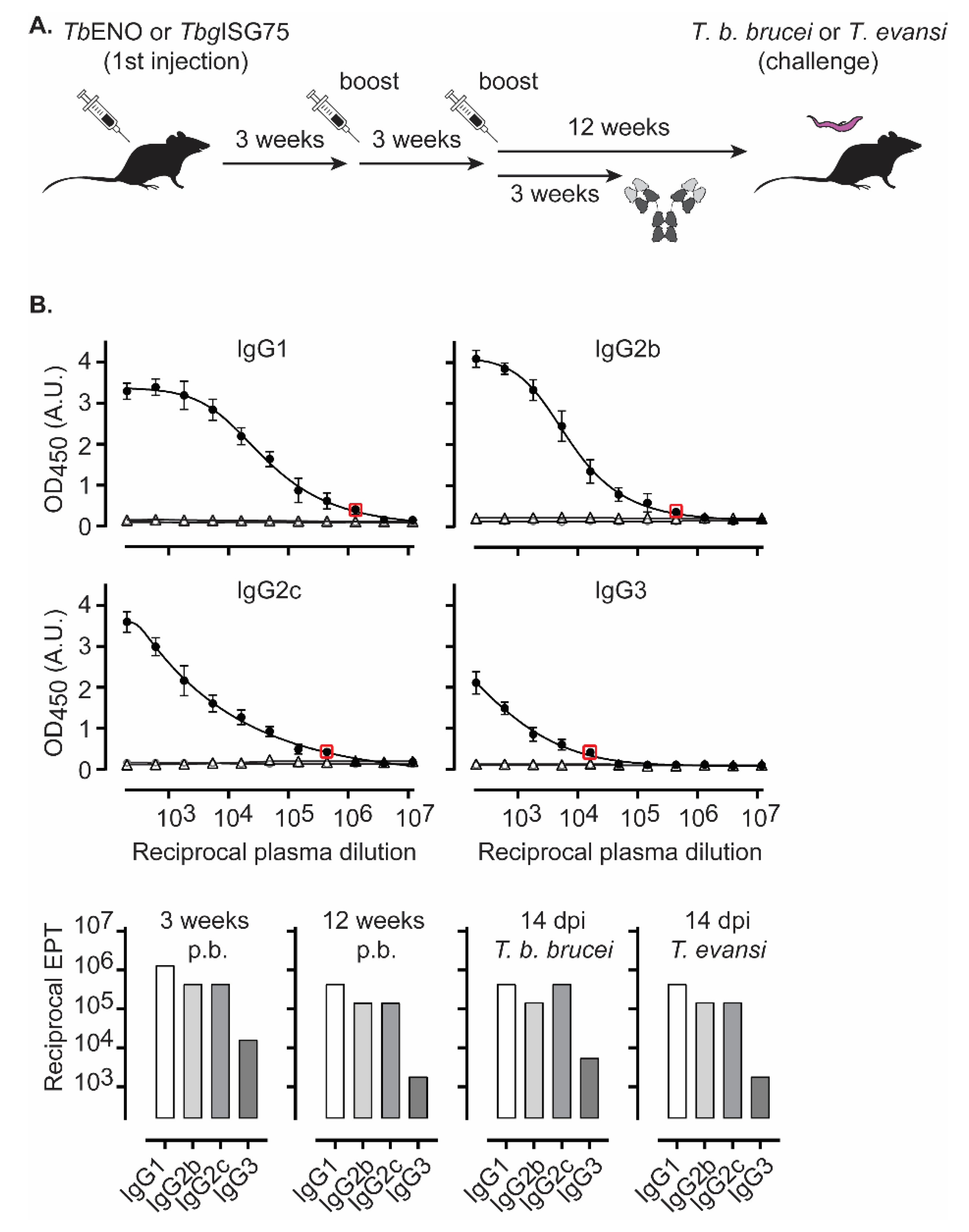
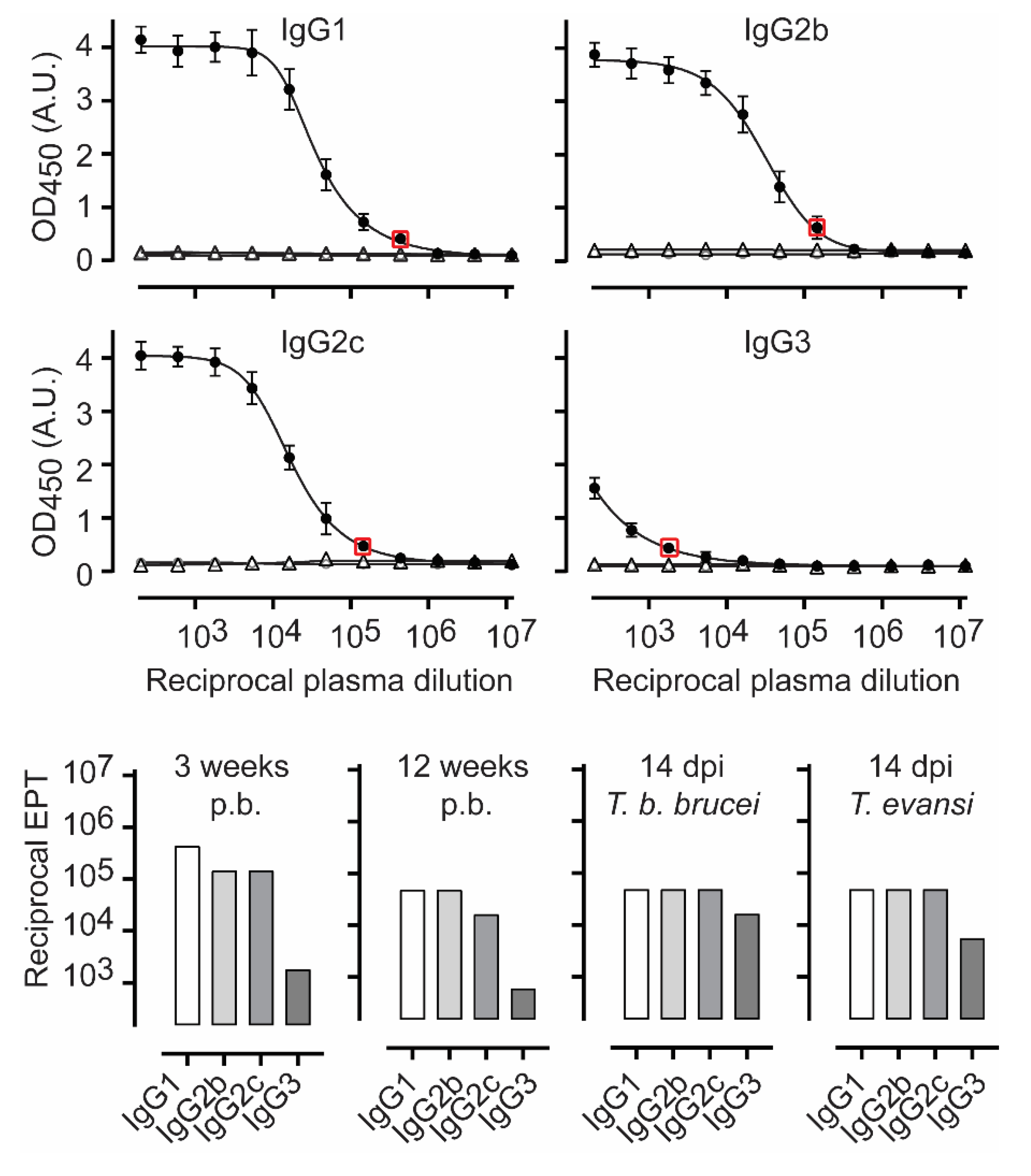
3.2. TbENO Is a Highly Immunogenic Antigen When Used in a CFA/IFA Vaccination Protocol
3.3. TbgISG75 Is a Highly Immunogenic Antigen When Used in a CFA/IFA Vaccination Protocol
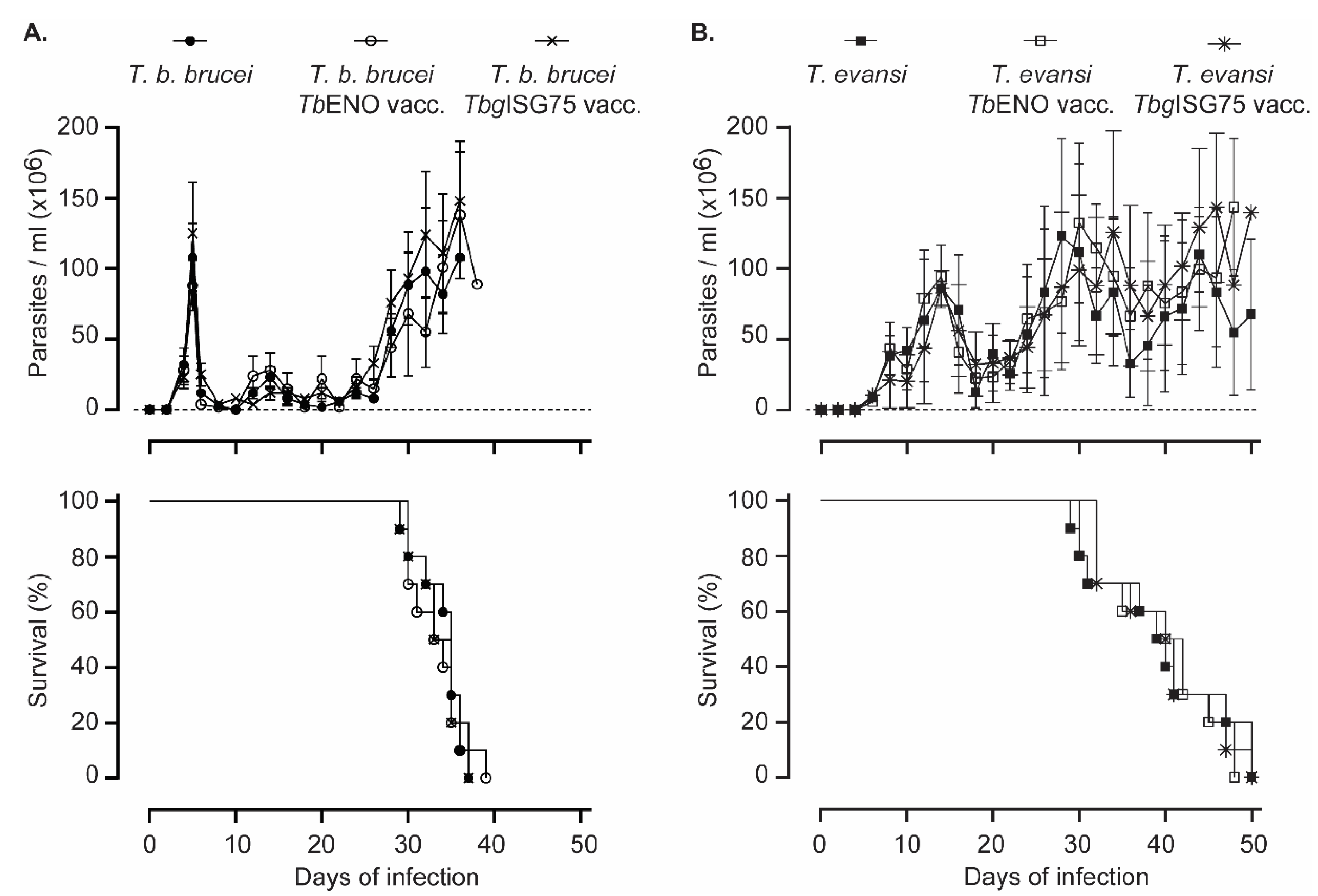
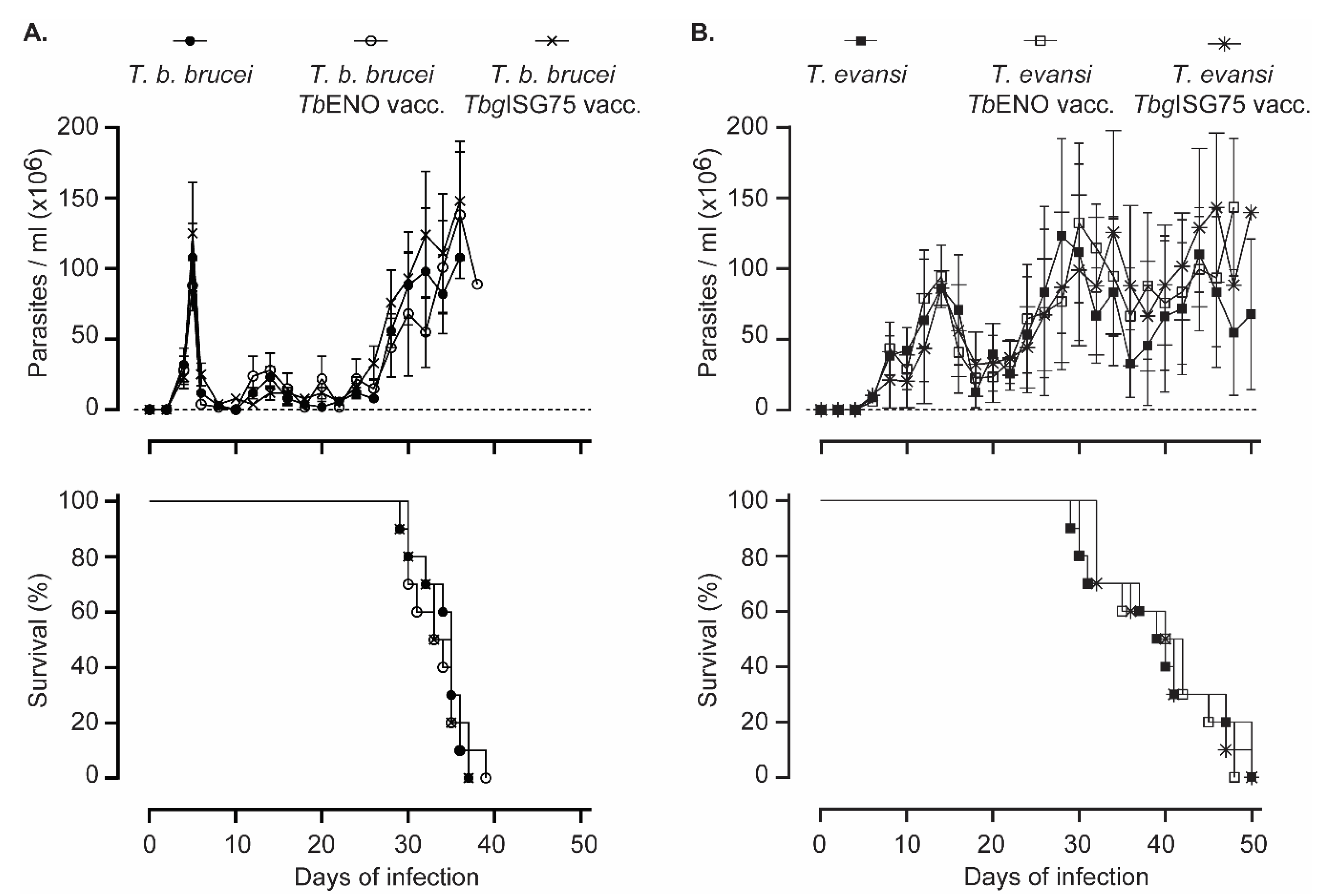
3.4. TbENO/TbgISG75 Vaccination Does Not Alter T. b. Brucei or T. evansi Infection Progression
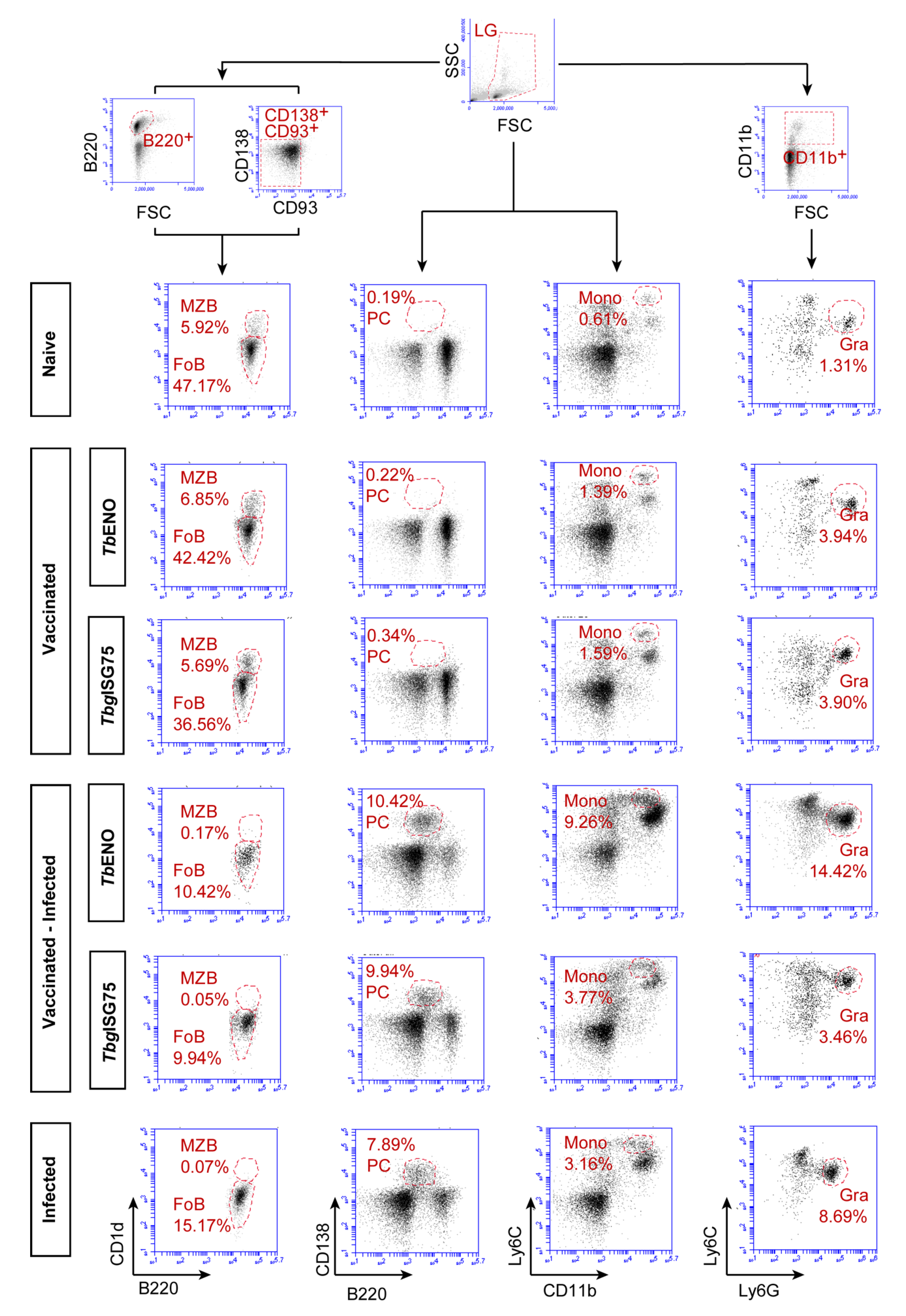
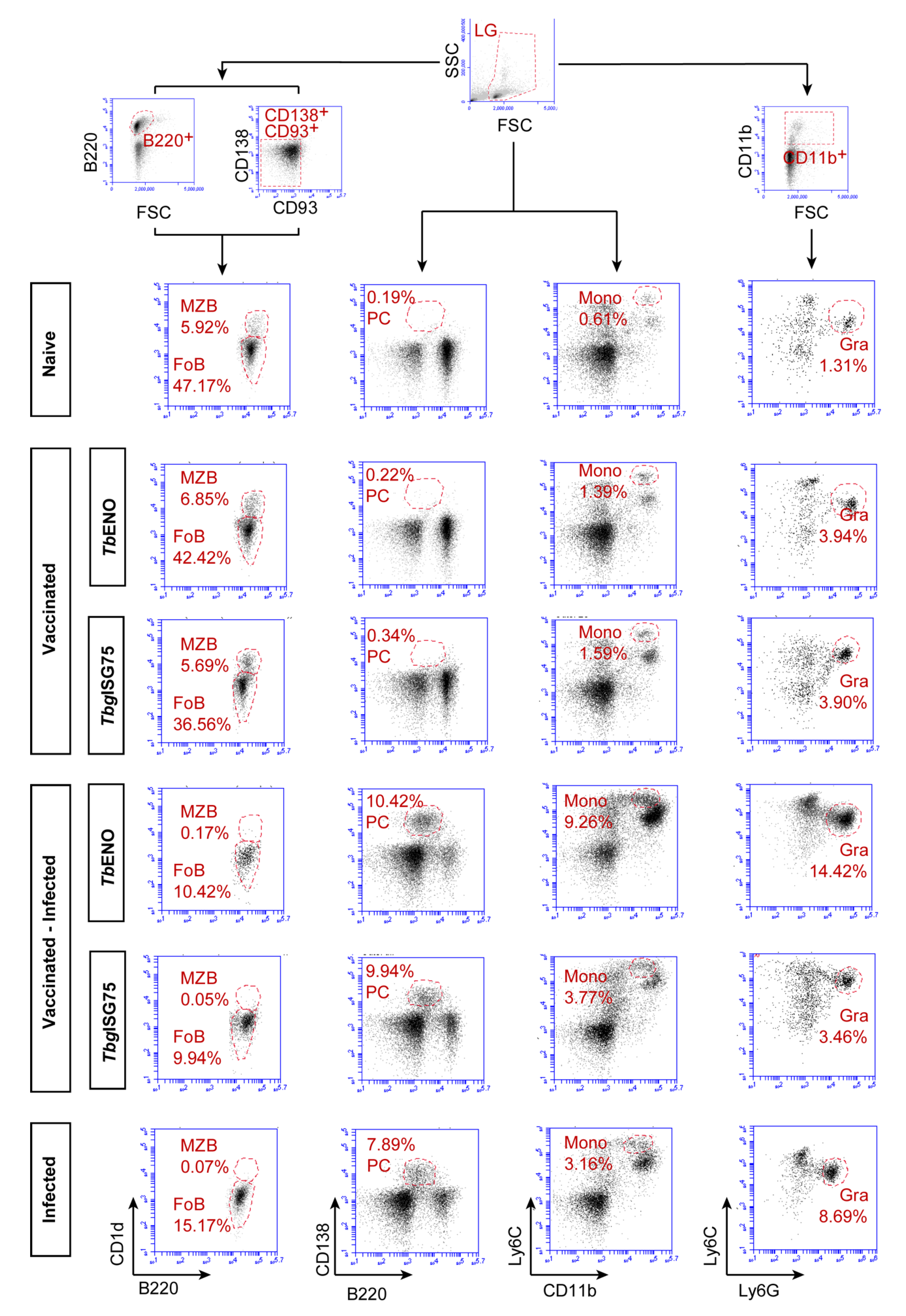
3.5. TbENO/TbgISG75 Vaccination Does Not Alter T. b. brucei Infection-Associated Immunopathology
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mogk, S.; Boßelmann, C.M.; Mudogo, C.N.; Stein, J.; Wolburg, H.; Duszenko, M. African trypanosomes and brain infection—The unsolved question. Biol. Rev. 2017, 92, 1675–1687. [Google Scholar] [CrossRef]
- Kristensson, K.; Nygard, M.; Bertini, G.; Bentivoglio, M. African trypanosome infections of the nervous system: Parasite entry and effects on sleep and synaptic functions. Prog. Neurobiol. 2010, 91, 152–171. [Google Scholar] [CrossRef]
- Trindade, S.; Rijo-Ferreira, F.; Carvalho, T.; Pinto-Neves, D.; Guegan, F.; Aresta-Branco, F.; Bento, F.; Young, S.A.; Pinto, A.; Abbeele, J.V.D.; et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 2016, 19, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, M.; Vereecke, N.; Deleeuw, V.; Pinto, J.; Magez, S. Salivarian Trypanosomosis: A Review of Parasites Involved, Their Global Distribution and Their Interaction with the Innate and Adaptive Mammalian Host Immune System. Front. Immunol. 2018, 9, 2253. [Google Scholar] [CrossRef] [PubMed]
- Capewell, P.; Cooper, A.; Clucas, C.; Weir, W.; Macleod, A. A co-evolutionary arms race: Trypanosomes shaping the human genome, humans shaping the trypanosome genome. Parasitology 2015, 142, S108–S119. [Google Scholar] [CrossRef] [Green Version]
- Pays, E.; Vanhollebeke, B.; Uzureau, P.; Lecordier, L.; Pérez-Morga, D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Microbiol. 2014, 12, 575–584. [Google Scholar] [CrossRef]
- Zoll, S.; Lane-Serff, H.; Mehmood, S.; Schneider, J.; Robinson, C.V.; Carrington, M.; Higgins, M.K. The structure of serum resistance-associated protein and its implications for human African trypanosomiasis. Nat. Microbiol. 2018, 17, 1–9. [Google Scholar] [CrossRef]
- Truc, P.; Büscher, P.; Cuny, G.; Gonzatti, M.I.; Jannin, J.; Joshi, P.; Juyal, P.; Lun, Z.-R.; Mattioli, R.; Pays, E.; et al. Atypical Human Infections by Animal Trypanosomes. PLoS Neglect. Trop. Dis. 2013, 7, e2256. [Google Scholar] [CrossRef] [Green Version]
- Chau, N.V.V.; Chau, L.B.; Desquesnes, M.; Herder, S.; Lan, N.P.H.; Campbell, J.I.; Cuong, N.V.; Yimming, B.; Chalermwong, P.; Jittapalapong, S.; et al. A Clinical and Epidemiological Investigation of the First Reported Human Infection with the Zoonotic Parasite Trypanosoma evansi in Southeast Asia. Clin. Infect. Dis 2016, 62, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Vanhollebeke, B.; Truc, P.; Poelvoorde, P.; Pays, A.; Joshi, P.P.; Katti, R.; Jannin, J.G.; Pays, E. Human Trypanosoma evansi Infection Linked to a Lack of Apolipoprotein L-I. N. Engl. J. Med. 2006, 355, 2752–2756. [Google Scholar] [CrossRef]
- Osório, A.L.A.R.; Madruga, C.R.; Desquesnes, M.; Soares, C.O.; Ribeiro, L.R.R.; Costa, S.C.G. da Trypanosoma (Duttonella) vivax: Its biology, epidemiology, pathogenesis, and introduction in the New World—A review. Memórias Inst. Oswaldo Cruz 2008, 103, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dyonisio, G.H.S.; Batista, H.R.; da Silva, R.E.; Azevedo, R.C.D.F.E.; Costa, J.D.O.J.; Manhes, I.B.D.O.; Tonhosolo, R.; Gennari, S.M.; Minervino, A.H.H.; Marcili, A. Molecular Diagnosis and Prevalence of Trypanosoma vivax (Trypanosomatida: Trypanosomatidae) in Buffaloes and Ectoparasites in the Brazilian Amazon Region. J. Med. Entomol. 2020, 58, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Larrea, M.A.; Medina-Pozo, M.L.; Cholota-Iza, C.E.; Jumbo-Moreira, J.R.; Saegerman, C.; Proaño-Pérez, F.; Ron-Román, J.; Reyna-Bello, A. First report and molecular identification of Trypanosoma (Duttonella) vivax outbreak in cattle population from Ecuador. Transbound. Emerg. Dis. 2020, 68, 2422–2428. [Google Scholar] [CrossRef] [PubMed]
- Desquesnes, M.; Holzmuller, P.; Lai, D.-H.; Dargantes, A.; Lun, Z.-R.; Jittaplapong, S. Trypanosoma evansi and Surra: A Review and Perspectives on Origin, History, Distribution, Taxonomy, Morphology, Hosts, and Pathogenic Effects. BioMed Res. Int. 2013, 2013, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Desquesnes, M.; Dargantes, A.; Lai, D.-H.; Lun, Z.-R.; Holzmuller, P.; Jittapalapong, S. Trypanosoma evansiand Surra: A Review and Perspectives on Transmission, Epidemiology and Control, Impact, and Zoonotic Aspects. BioMed Res. Int. 2013, 2013, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, C.; Desquesnes, M.; Touratier, L.; Büscher, P. Trypanosoma evansi: Recent outbreaks in Europe. Vet. Parasitol. 2010, 174, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, A.; Gutierrez, C.; Arroyo, R.; Jimenez, V.; Zagalá, G.; Bosch, I.; Sirvent, J.; Alberola, J.; Alonso, I.; Caballero, C. Trypanosoma evansi infection in mainland Spain. Vet. Parasitol. 2010, 167, 74–76. [Google Scholar] [CrossRef]
- Defontis, M.; Richartz, J.; Engelmann, N.; Bauer, C.; Schwierk, V.M.; Büscher, P.; Moritz, A. Canine Trypanosoma evansi infection introduced into Germany. Vet. Clin. Path. 2012, 41, 369–374. [Google Scholar] [CrossRef]
- Desquesnes, M.; Bossard, G.; Patrel, D.; Herder, S.; Patout, O.; Lepetitcolin, E.; Thevenon, S.; Berthier, D.; Pavlovic, D.; Brugidou, R.; et al. First outbreak of Trypanosoma evansi in camels in metropolitan France. Vet. Rec. 2008, 162, 750–752. [Google Scholar] [CrossRef]
- Rodríguez, N.F.; Tejedor-Junco, M.T.; González-Martín, M.; Gutierrez, C. Stomoxys calcitrans as possible vector of Trypanosoma evansi among camels in an affected area of the Canary Islands, Spain. Rev. Soc. Bras. Med. Trop. 2014, 47, 510–512. [Google Scholar] [CrossRef]
- Baldacchino, F.; Desquesnes, M.; Mihok, S.; Foil, L.D.; Duvallet, G.; Jittapalapong, S. Tabanids: Neglected subjects of research, but important vectors of disease agents! Infect. Genet. Evol. 2014, 28, 596–615. [Google Scholar] [CrossRef] [PubMed]
- Aregawi, W.G.; Agga, G.E.; Abdi, R.D.; Büscher, P. Systematic review and meta-analysis on the global distribution, host range, and prevalence of Trypanosoma evansi. Parasite Vector 2019, 12, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magez, S.; Torres, J.E.P.; Oh, S.; Radwanska, M. Salivarian Trypanosomes Have Adopted Intricate Host-Pathogen Interaction Mechanisms That Ensure Survival in Plain Sight of the Adaptive Immune System. Pathogens 2021, 10, 679. [Google Scholar] [CrossRef]
- Bangs, J.D. Evolution of Antigenic Variation in African Trypanosomes: Variant Surface Glycoprotein Expression, Structure, and Function. Bioessays 2018, 40, 1800181. [Google Scholar] [CrossRef] [PubMed]
- Schwede, A.; Macleod, O.J.S.; MacGregor, P.; Carrington, M. How Does the VSG Coat of Bloodstream Form African Trypanosomes Interact with External Proteins? PLoS Pathog. 2015, 11, e1005259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugnier, M.R.; Cross, G.A.; Papavasiliou, N. The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science 2015, 347, 1470–1473. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, R.; Cobbold, C.A.; Figueiredo, L.; Jackson, A.; Morrison, L.J.; Mugnier, M.R.; Papavasiliou, N.; Schnaufer, A.; Matthews, K. Emerging challenges in understanding trypanosome antigenic variation. Emerg. Top. Life Sci. 2017, 1, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Pinger, J.; Nešić, D.; Ali, L.; Aresta-Branco, F.; Lilic, M.; Chowdhury, S.; Kim, H.-S.; Verdi, J.; Raper, J.; Ferguson, M.A.J.; et al. African trypanosomes evade immune clearance by O-glycosylation of the VSG surface coat. Nat. Microbiol. 2018, 3, 932–938. [Google Scholar] [CrossRef]
- Engstler, M.; Pfohl, T.; Herminghaus, S.; Boshart, M.; Wiegertjes, G.; Heddergott, N.; Overath, P. Hydrodynamic Flow-Mediated Protein Sorting on the Cell Surface of Trypanosomes. Cell 2007, 131, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Dean, S.D.; Matthews, K.R. Restless Gossamers: Antibody Clearance by Hydrodynamic Flow Forces Generated at the Surface of Motile Trypanosome Parasites. Cell Host Microbe 2007, 2, 279–281. [Google Scholar] [CrossRef] [Green Version]
- Devine, D.V.; Falk, R.J.; Balber, A.E. Restriction of the alternative pathway of human complement by intact Trypanosoma brucei subsp. gambiense. Infect. Immun. 1986, 52, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinger, J.; Chowdhury, S.; Papavasiliou, F.N. Variant surface glycoprotein density defines an immune evasion threshold for African trypanosomes undergoing antigenic variation. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Ogunremi, O.; Wei, G.; Shi, M.; Tabel, H. CR3 (CD11b/CD18) is the major macrophage receptor for IgM antibody-mediated phagocytosis of African trypanosomes: Diverse effect on subsequent synthesis of tumor necrosis factor α and nitric oxide. Microbes Infect. 2006, 8, 1209–1218. [Google Scholar] [CrossRef]
- Liu, G.; Fu, Y.; Yosri, M.; Chen, Y.; Sun, P.; Xu, J.; Zhang, M.; Sun, D.; Strickland, A.B.; Mackey, Z.B.; et al. CRIg plays an essential role in intravascular clearance of bloodborne parasites by interacting with complement. Proc. Natl. Acad. Sci. USA 2019, 116, 24214–24220. [Google Scholar] [CrossRef]
- Dagenais, T.R.; Demick, K.P.; Bangs, J.D.; Forest, K.T.; Paulnock, D.M.; Mansfield, J.M. T-Cell Responses to the Trypanosome Variant Surface Glycoprotein Are Not Limited to Hypervariable Subregions. Infect. Immun. 2009, 77, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, D.; Zhang, F.; Guirnalda, P.; Haynes, C.; Bockstal, V.; Radwanska, M.; Magez, S.; Black, S.J. Trypanosoma brucei Co-opts NK Cells to Kill Splenic B2 B Cells. PLoS Pathog. 2016, 12, e1005733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockstal, V.; Guirnalda, P.; Caljon, G.; Goenka, R.; Telfer, J.C.; Frenkel, D.; Radwanska, M.; Magez, S.; Black, S.J. T. brucei Infection Reduces B Lymphopoiesis in Bone Marrow and Truncates Compensatory Splenic Lymphopoiesis through Transitional B-Cell Apoptosis. PLoS Pathog. 2011, 7, e1002089. [Google Scholar] [CrossRef] [Green Version]
- Obishakin, E.; Trez, C.; Magez, S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol. 2014, 36, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Blom-Potar, M.C.; Chamond, N.; Cosson, A.; Jouvion, G.; Droin-Bergère, S.; Huerre, M.; Minoprio, P. Trypanosoma vivax Infections: Pushing Ahead with Mouse Models for the Study of Nagana. II. Immunobiological Dysfunctions. PLoS Neglect. Trop. Dis. 2010, 4, e793. [Google Scholar] [CrossRef] [Green Version]
- Radwanska, M.; Guirnalda, P.; Trez, C.D.; Ryffel, B.; Black, S.; Magez, S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008, 4, e1000078. [Google Scholar] [CrossRef] [Green Version]
- Magez, S.; Schwegmann, A.; Atkinson, R.; Claes, F.; Drennan, M.; Baetselier, P.D.; Brombacher, F. The Role of B-cells and IgM Antibodies in Parasitemia, Anemia, and VSG Switching in Trypanosoma brucei–Infected Mice. PLoS Pathog. 2008, 4, e1000122. [Google Scholar] [CrossRef] [Green Version]
- McNae, I.W.; Kinkead, J.; Malik, D.; Yen, L.-H.; Walker, M.K.; Swain, C.; Webster, S.P.; Gray, N.; Fernandes, P.M.; Myburgh, E.; et al. Fast acting allosteric phosphofructokinase inhibitors block trypanosome glycolysis and cure acute African trypanosomiasis in mice. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Borst, P. Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: The glycosome. Febs Lett. 1977, 80, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Visser, N.; Opperdoes, F.R. Glycolysis in Trypanosoma brucei. Eur. J. Biochem. 1980, 103, 623–632. [Google Scholar] [CrossRef]
- Szöör, B.; Haanstra, J.R.; Gualdrón-López, M.; Michels, P.A. Evolution, dynamics and specialized functions of glycosomes in metabolism and development of trypanosomatids. Curr. Opin. Microbiol. 2014, 22, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haanstra, J.R.; González-Marcano, E.B.; Gualdrón-López, M.; Michels, P.A. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochim. Biophys. Acta 2016, 1863, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.B.; Lee, K.-Y.; Mireji, P.; Enyaru, J.; Sistrom, M.; Aksoy, S.; Zhao, H.; Caccone, A. Genomic analyses of African Trypanozoon strains to assess evolutionary relationships and identify markers for strain identification. PLoS Neglect. Trop. Dis. 2017, 11, e0005949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.A.; Nava, M. Trypanosoma evansi is alike to Trypanosoma brucei brucei in the subcellular localisation of glycolytic enzymes. Memórias Inst. Oswaldo Cruz 2015, 110, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Rivero, L.A.; Concepción, J.L.; Quintero-Troconis, E.; Quiñones, W.; Michels, P.A.M.; Acosta, H. Trypanosoma evansi contains two auxiliary enzymes of glycolytic metabolism: Phosphoenolpyruvate carboxykinase and pyruvate phosphate dikinase. Exp. Parasitol. 2016, 165, 7–15. [Google Scholar] [CrossRef]
- Hannaert, V.; Albert, M.-A.; Rigden, D.J.; Giotto, M.T.D.S.; Thiemann, O.; Garratt, R.C.; Roy, J.V.; Opperdoes, F.R.; Michels, P.A. Kinetic characterization, structure modelling studies and crystallization of Trypanosoma brucei enolase. Eur. J. Biochem. 2003, 270, 3205–3213. [Google Scholar] [CrossRef]
- Giotto, M.T.D.S.; Hannaert, V.; Vertommen, D.; Navarro, M.V.D.A.S.; Rider, M.H.; Michels, P.A.; Garratt, R.C.; Rigden, D.J. The Crystal Structure of Trypanosoma brucei Enolase: Visualisation of the Inhibitory Metal Binding Site III and Potential as Target for Selective, Irreversible Inhibition. J. Mol. Biol. 2003, 331, 653–665. [Google Scholar] [CrossRef]
- Navarro, M.V.D.A.S.; Dias, S.M.G.; Mello, L.V.; Giotto, M.T.D.S.; Gavalda, S.; Blonski, C.; Garratt, R.C.; Rigden, D.J. Structural flexibility in Trypanosoma brucei enolase revealed by X-ray crystallography and molecular dynamics. FEBS J. 2007, 274, 5077–5089. [Google Scholar] [CrossRef] [PubMed]
- Grébaut, P.; Chuchana, P.; Brizard, J.-P.; Demettre, E.; Seveno, M.; Bossard, G.; Jouin, P.; Vincendeau, P.; Bengaly, Z.; Boulangé, A.; et al. Identification of total and differentially expressed excreted–secreted proteins from Trypanosoma congolense strains exhibiting different virulence and pathogenicity. Int. J. Parasitol. 2009, 39, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Geiger, A.; Hirtz, C.; Bécue, T.; Bellard, E.; Centeno, D.; Gargani, D.; Rossignol, M.; Cuny, G.; Peltier, J.-B. Exocytosis and protein secretion in Trypanosoma. BMC Microbiol. 2010, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nten, C.M.A.; Sommerer, N.; Rofidal, V.; Hirtz, C.; Rossignol, M.; Cuny, G.; Peltier, J.-B.; Geiger, A. Excreted/Secreted Proteins from Trypanosome Procyclic Strains. J. Biomed. Biotechnol. 2010, 2010, 212817. [Google Scholar] [CrossRef] [Green Version]
- Szempruch, A.J.; Sykes, S.E.; Kieft, R.; Dennison, L.; Becker, A.C.; Gartrell, A.; Martin, W.J.; Nakayasu, E.S.; Almeida, I.C.; Hajduk, S.L.; et al. Extracellular Vesicles from Trypanosoma brucei Mediate Virulence Factor Transfer and Cause Host Anemia. Cell 2016, 164, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Torres, J.E.P.; Goossens, J.; Vertommen, D.; Caljon, G.; Sterckx, Y.G.J.; Magez, S. An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi. Vaccines 2020, 8, 415. [Google Scholar] [CrossRef]
- Avilán, L.; Gualdrón-López, M.; Quiñones, W.; González-González, L.; Hannaert, V.; Michels, P.A.; Concepción, J.L. Enolase: A key player in the metabolism and a probable virulence factor of trypanosomatid parasites-perspectives for its use as a therapeutic target. Enzyme Res. 2011, 2011, 932549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiñones, W.; Peña, P.; Domingo-Sananes, M.; Cáceres, A.; Michels, P.A.M.; Avilan, L.; Concepción, J.L. Leishmania mexicana: Molecular cloning and characterization of enolase. Exp. Parasitol. 2007, 116, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Vanegas, G.; Quiñones, W.; Carrasco-López, C.; Concepción, J.L.; Albericio, F.; Avilán, L. Enolase as a plasminogen binding protein in Leishmania mexicana. Parasitol. Res. 2007, 101, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemar, N.; Zwyer, M.; Zoltner, M.; Cal, M.; Field, M.C.; Mäser, P. Expression of a specific variant surface glycoprotein has a major impact on suramin sensitivity and endocytosis in Trypanosoma brucei. Faseb Bioadv. 2019, 1, 595–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoltner, M.; Campagnaro, G.D.; Taleva, G.; Burrell, A.; Cerone, M.; Leung, K.F.; Achcar, F.; Horn, D.; Vaughan, S.; Gadelha, C.; et al. Suramin exposure alters cellular metabolism and mitochondrial energy production in African trypanosomes. J. Biol. Chem. 2020, 295, 8331–8347. [Google Scholar] [CrossRef]
- Ziegelbauer, K.; Overath, P. Organization of two invariant surface glycoproteins in the surface coat of Trypanosoma brucei. Infect. Immun. 1993, 61, 4540–4545. [Google Scholar] [CrossRef] [Green Version]
- Ziegelbauer, K.; Overath, P. Identification of invariant surface glycoproteins in the bloodstream stage of Trypanosoma brucei. J. Biol. Chem. 1992, 267, 10791–10796. [Google Scholar] [CrossRef]
- Ziegelbauer, K.; Multhaup, G.; Overath, P. Molecular characterization of two invariant surface glycoproteins specific for the bloodstream stage of Trypanosoma brucei. J. Biol. Chem. 1992, 267, 10797–10803. [Google Scholar] [CrossRef]
- Radwanska, M.; Magez, S.; Michel, A.; Stijlemans, B.; Geuskens, M.; Pays, E. Comparative Analysis of Antibody Responses against HSP60, Invariant Surface Glycoprotein 70, and Variant Surface Glycoprotein Reveals a Complex Antigen-Specific Pattern of Immunoglobulin Isotype Switching during Infection by Trypanosoma brucei. Infect. Immun. 2000, 68, 848–860. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.; Wall, S.J.; Carrington, M.; Ferguson, M.A.J. Proteomic Selection of Immunodiagnostic Antigens for Human African Trypanosomiasis and Generation of a Prototype Lateral Flow Immunodiagnostic Device. PLoS Neglect. Trop. Dis. 2013, 7, e2087. [Google Scholar] [CrossRef] [PubMed]
- Biéler, S.; Waltenberger, H.; Barrett, M.P.; McCulloch, R.; Mottram, J.C.; Carrington, M.; Schwaeble, W.; McKerrow, J.; Phillips, M.A.; Michels, P.A.; et al. Evaluation of Antigens for Development of a Serological Test for Human African Trypanosomiasis. PLoS ONE 2016, 11, e0168074. [Google Scholar] [CrossRef] [Green Version]
- Rudramurthy, G.R.; Sengupta, P.P.; Metilda, B.; Balamurugan, V.; Prabhudas, K.; Rahman, H. Development of an enzyme immunoassay using recombinant invariant surface glycoprotein (rISG) 75 for serodiagnosis of bovine trypanosomosis. Indian J. Exp. Biol. 2015, 53, 7–15. [Google Scholar]
- Rudramurthy, G.R.; Sengupta, P.P.; Ligi, M.; Rahman, H. An inhibition enzyme immuno assay exploring recombinant invariant surface glycoprotein and monoclonal antibodies for surveillance of surra in animals. Biologicals 2017, 46, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Koumandou, V.L.; Boehm, C.; Horder, K.A.; Field, M.C. Evidence for Recycling of Invariant Surface Transmembrane Domain Proteins in African Trypanosomes. Eukaryot. Cell 2013, 12, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.F.; Riley, F.S.; Carrington, M.; Field, M.C. Ubiquitylation and Developmental Regulation of Invariant Surface Protein Expression in Trypanosomes. Eukaryot. Cell 2011, 10, 916–931. [Google Scholar] [CrossRef] [Green Version]
- Radwanska, M.; Magez, S.; Dumont, N.; Pays, A.; Nolan, D.; Pays, E. Antibodies raised against the flagellar pocket fraction of Trypanosoma brucei preferentially recognize HSP60 in cDNA expression library. Parasite Immunol. 2000, 22, 639–650. [Google Scholar] [CrossRef]
- Aricescu, A.R.; Lu, W.; Jones, E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 1243–1250. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Hajizadeh, N.R.; Franke, D.; Jeffries, C.M.; Svergun, D.I. Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009, 42, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2020, 30, 70–82. [Google Scholar] [CrossRef]
- Deleeuw, V.; Phạm, H.T.T.; Poorter, I.D.; Janssens, I.; Trez, C.D.; Radwanska, M.; Magez, S. Trypanosoma brucei brucei causes a rapid and persistent influx of neutrophils in the spleen of infected mice. Parasite Immunol. 2019, 41, e12664. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.K.; Carrington, M. Sequence variation and structural conservation allows development of novel function and immune evasion in parasite surface protein families. Protein Sci. 2014, 23, 354–365. [Google Scholar] [CrossRef]
- Blum, M.; Down, J.; Gurnett, A.; Carrington, M.; Turner, M.; Wiley, D. A structural motif in the variant surface glycoproteins of Trypanosoma brucei. Nature 1993, 362, 603–609. [Google Scholar] [CrossRef]
- Trevor, C.E.; Gonzalez-Munoz, A.L.; Macleod, O.J.S.; Woodcock, P.G.; Rust, S.; Vaughan, T.J.; Garman, E.F.; Minter, R.; Carrington, M.; Higgins, M.K. Structure of the trypanosome transferrin receptor reveals mechanisms of ligand recognition and immune evasion. Nat. Microbiol. 2019, 4, 2074–2081. [Google Scholar] [CrossRef]
- Bartossek, T.; Jones, N.G.; Schäfer, C.S.; Cvitković, M.C.; Glogger, M.; Mott, H.R.; Kuper, J.; Brennich, M.; Carrington, M.; Smith, A.-S.; et al. Structural basis for the shielding function of the dynamic trypanosome variant surface glycoprotein coat. Nat. Microbiol. 2017, 2, 1523–1532. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.K.; Tkachenko, O.; Brown, A.; Reed, J.; Raper, J.; Carrington, M. Structure of the trypanosome haptoglobin-hemoglobin receptor and implications for nutrient uptake and innate immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 1905–1910. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Omer, S.B. Why and How Vaccines Work. Cell 2020, 183, 290–295. [Google Scholar] [CrossRef]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, J.A.; Lo, N.C.; Arinaminpathy, N.; Frost, I.; Laxminarayan, R. Childhood vaccines and antibiotic use in low- and middle-income countries. Nature 2020, 581, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Dyes, trypanosomiasis and DNA: A historical and critical review. Biotech. Histochem. 2010, 85, 341–354. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Cerami, A. Hypertriglyceridemia associated with Trypanosoma brucei brucei infection in rabbits: Role of defective triglyceride removal. Mol. Biochem. Parasit. 1980, 2, 31–38. [Google Scholar] [CrossRef]
- Beutler, B.; Cerami, A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 1986, 320, 584–588. [Google Scholar] [CrossRef]
- Cross, G.A.M. Antigenic variation in trypanosomes. Proc. Royal. Soc. Lond. Ser. B Biol. Sci. 1978, 202, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Cross, G.A.M. Identification, purification and properties of clone-specific glycoprotein antigens constituting the surface coat of Trypanosoma brucei. Parasitology 1975, 71, 393–417. [Google Scholar] [CrossRef]
- Vickerman, K. Antigenic variation in trypanosomes. Nature 1978, 273, 613–617. [Google Scholar] [CrossRef]
- Cornelissen, A.W.C.A.; Bakkeren, G.A.M.; Barry, J.D.; Michels, P.A.M.; Borst, P. Characteristics of trypanosome variant antigen genes active in the tsetse fly. Nucleic Acids Res. 1985, 13, 4661–4676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzcanga, G.L.; Perrone, T.; Noda, J.A.; Pérez-Pazos, J.; Medina, R.; Hoebeke, J.; Bubis, J. Variant Surface Glycoprotein from Trypanosoma evansi Is Partially Responsible for the Cross-Reaction between Trypanosoma evansi and Trypanosoma vivax. Biochemistry 2004, 43, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Magez, S.; Torres, J.E.P.; Obishakin, E.; Radwanska, M. Infections with Extracellular Trypanosomes Require Control by Efficient Innate Immune Mechanisms and Can Result in the Destruction of the Mammalian Humoral Immune System. Front. Immunol. 2020, 11, 382. [Google Scholar] [CrossRef] [Green Version]
- Radwanska, M.; Nguyen, H.T.T.; Moon, S.; Obishakin, E.; Magez, S. Trypanosomatids, Methods and Protocols. Methods Mol. Biol. 2020, 2116, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Maharana, B.R.; Sudhakar, N.R.; Jawalagatti, V.; Saravanan, B.C.; Blake, D.P.; Tewari, A.K. Evaluation of the Immunoprotective Potential of Recombinant Paraflagellar Rod Proteins of Trypanosoma evansi in Mice. Vaccines 2020, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autheman, D.; Crosnier, C.; Clare, S.; Goulding, D.A.; Brandt, C.; Harcourt, K.; Tolley, C.; Galaway, F.; Khushu, M.; Ong, H.; et al. An invariant Trypanosoma vivax vaccine antigen induces protective immunity. Nature 2021, 595, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mkunza, F.; Olaho, W.M.; Powell, C.N. Partial protection against natural trypanosomiasis after vaccination with a flagellar pocket antigen from Trypanosoma brucei rhodesiense. Vaccine 1995, 13, 151–154. [Google Scholar] [CrossRef]
- Southon, H.A.W.; Cunningham, M.P. Infectivity of Trypanosomes derived from Individual Glossina morsitans Westw. Nature 1966, 212, 1477–1478. [Google Scholar] [CrossRef]
- Russo, D.C.; Grab, D.J.; Lonsdale-Eccles, J.D.; Shaw, M.K.; Williams, D.J. Directional movement of variable surface glycoprotein-antibody complexes in Trypanosoma brucei. Eur. J. Cell Biol. 1993, 62, 432–441. [Google Scholar]
- Dickie, E.A.; Giordani, F.; Gould, M.K.; Mäser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Infect. Dis. 2020, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Guedes, R.L.M.; Rodrigues, C.M.F.; Coatnoan, N.; Cosson, A.; Cadioli, F.A.; Garcia, H.A.; Gerber, A.L.; Machado, R.Z.; Minoprio, P.M.C.; Teixeira, M.M.G.; et al. A comparative in silico linear B-cell epitope prediction and characterization for South American and African Trypanosoma vivax strains. Genomics 2019, 111, 407–417. [Google Scholar] [CrossRef]
- Liu, T.; Shi, K.; Li, W. Deep learning methods improve linear B-cell epitope prediction. Biodata Min. 2020, 13, 1–3. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magez, S.; Li, Z.; Nguyen, H.T.T.; Pinto Torres, J.E.; Van Wielendaele, P.; Radwanska, M.; Began, J.; Zoll, S.; Sterckx, Y.G.-J. The History of Anti-Trypanosome Vaccine Development Shows That Highly Immunogenic and Exposed Pathogen-Derived Antigens Are Not Necessarily Good Target Candidates: Enolase and ISG75 as Examples. Pathogens 2021, 10, 1050. https://doi.org/10.3390/pathogens10081050
Magez S, Li Z, Nguyen HTT, Pinto Torres JE, Van Wielendaele P, Radwanska M, Began J, Zoll S, Sterckx YG-J. The History of Anti-Trypanosome Vaccine Development Shows That Highly Immunogenic and Exposed Pathogen-Derived Antigens Are Not Necessarily Good Target Candidates: Enolase and ISG75 as Examples. Pathogens. 2021; 10(8):1050. https://doi.org/10.3390/pathogens10081050
Chicago/Turabian StyleMagez, Stefan, Zeng Li, Hang Thi Thu Nguyen, Joar Esteban Pinto Torres, Pieter Van Wielendaele, Magdalena Radwanska, Jakub Began, Sebastian Zoll, and Yann G.-J. Sterckx. 2021. "The History of Anti-Trypanosome Vaccine Development Shows That Highly Immunogenic and Exposed Pathogen-Derived Antigens Are Not Necessarily Good Target Candidates: Enolase and ISG75 as Examples" Pathogens 10, no. 8: 1050. https://doi.org/10.3390/pathogens10081050
APA StyleMagez, S., Li, Z., Nguyen, H. T. T., Pinto Torres, J. E., Van Wielendaele, P., Radwanska, M., Began, J., Zoll, S., & Sterckx, Y. G.-J. (2021). The History of Anti-Trypanosome Vaccine Development Shows That Highly Immunogenic and Exposed Pathogen-Derived Antigens Are Not Necessarily Good Target Candidates: Enolase and ISG75 as Examples. Pathogens, 10(8), 1050. https://doi.org/10.3390/pathogens10081050