
Immunodominant Cytomegalovirus Epitopes Suppress Subdominant Epitopes in the Generation of High-Avidity CD8 T Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice, Viruses, and Infection Procedures
2.2. Preparation of Single-Cell Suspensions from Lungs and Spleen
2.3. Peptides and Quantitation of Functional Epitope-Specific CD8+ T Cells
2.4. Cytofluorometric Analyses
2.5. Statistics and Determination of Avidity Distributions
3. Results
3.1. Long-Term Kinetics of CD8+ T-Cell Memory during Latent Infection after Local Priming
3.2. Impact of IDEs on the Long-Term Kinetics of CD8+ T-Cell Memory Directed against Non-IDE m18
3.3. Functional Avidities of CD8+ T Cells Specific for IDEs
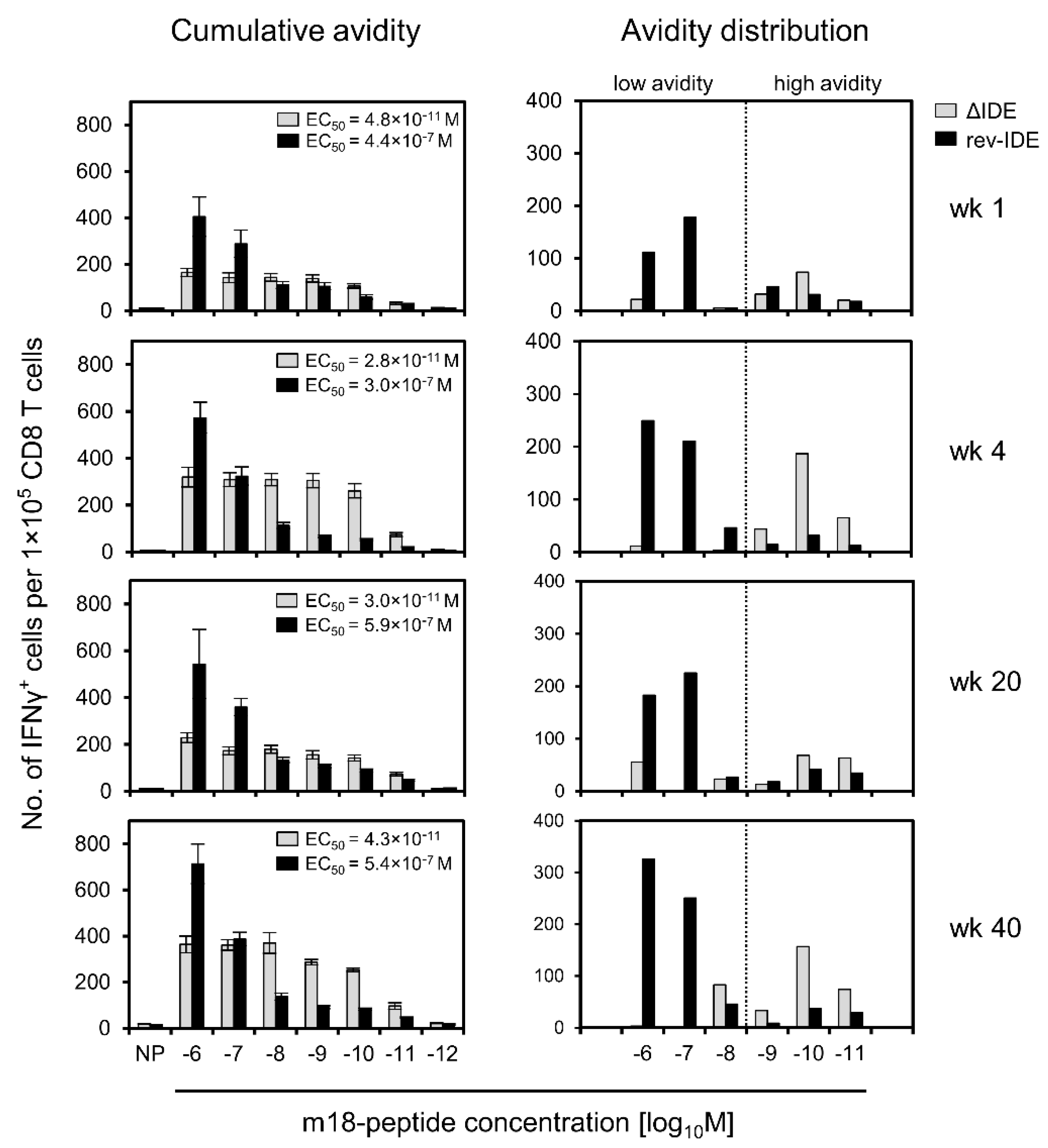
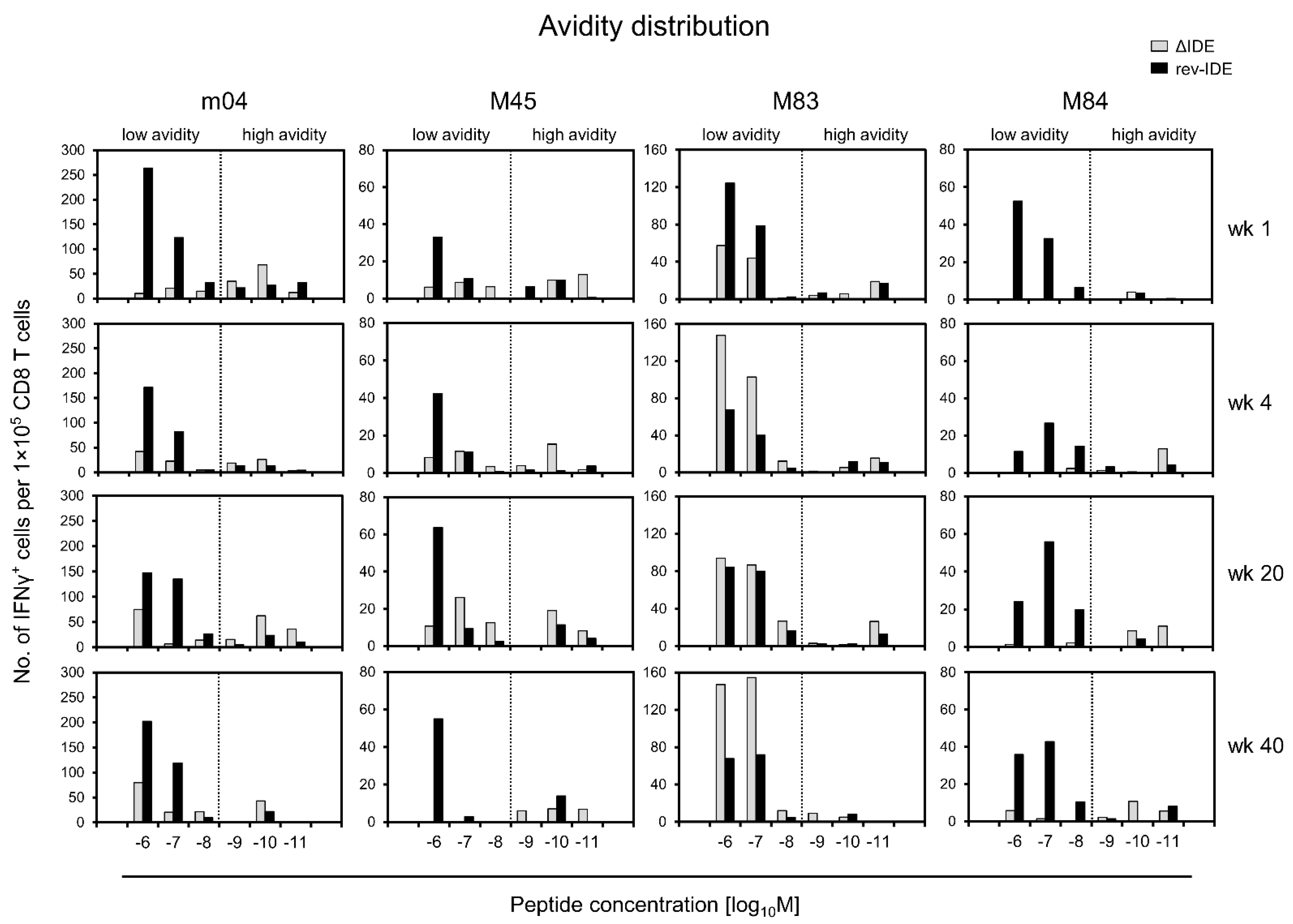
3.4. Absence of IDEs Facilitates Primarily the Expansion of High-Avidity CD8+ T Cells Specific for Non-IDEs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Roizman, B.; Sears, A.E. An inquiry into the mechanisms of herpes simplex virus latency. Annu. Rev. Microbiol. 1987, 41, 543–571. [Google Scholar] [CrossRef]
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 1–22. [Google Scholar]
- Redwood, A.J.; Shellam, G.R.; Smith, L.M. Molecular evolution of murine cytomegalovirus genomes. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 23–37. [Google Scholar]
- Cannon, M.J.; Grosse, S.D.; Fowler, K.B. The epidemiology and public health impact of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 26–48. [Google Scholar]
- Adler, S.P.; Nigro, G. Clinical cytomegalovirus research: Congential infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 55–73. [Google Scholar]
- Boppana, S.B.; Britt, W.J. Synopsis of clinical aspects of human cytomegalovirus disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 1–25. [Google Scholar]
- Taylor-Wiedeman, J.; Sissons, J.G.P.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; Bruening, E.E.; Donahue, R.E.; Mocarski, E.S.; Young, N.S.; St Jeor, S.C. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 1992, 80, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C.; Fish, K.; Nelson, J.A. Reactivation of Latent Human Cytomegalovirus by Allogeneic Stimulation of Blood Cells from Healthy Donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [Green Version]
- Söderberg-Nauclér, C.; Streblow, D.N.; Fish, K.; Allan-Yorke, J.; Smith, P.P.; Nelson, J.A. Reactivation of Latent Human Cytomegalovirus in CD14 + Monocytes Is Differentiation Dependent. J. Virol. 2001, 75, 7543–7554. [Google Scholar] [CrossRef] [Green Version]
- Hebart, H.; Einsele, H. Clinical aspects of CMV infection after stem cell transplantation. Hum. Immunol. 2004, 65, 432–436. [Google Scholar] [CrossRef]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Hematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 337–353. [Google Scholar]
- Reddehase, M.J.; Lemmermann, N.A.W. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef]
- Riddell, S.R. Pathogenesis of cytomegalovirus pneumonia in immunocompromised hosts. Semin. Respir. Infect. 1995, 10, 199–208. [Google Scholar] [PubMed]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human Cytomegalovirus Latency and Reactivation in Allogeneic Hematopoietic Stem Cell Transplant Recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffini, E.; Giaccone, L.; Festuccia, M.; Brunello, L.; Busca, A.; Bruno, B. Treatment of CMV Infection after Allogeneic Hematopoietic Stem Cell Transplantation. Expert Rev. Hematol. 2016, 9, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, R.K. Clinical cytomegaloviurs resarch: Thoracic organ transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 286–300. [Google Scholar]
- Emery, V.C.; Milne, R.S.B.; Griffiths, P.D. Clinical cytomegalovirus research: Liver and kidney transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 301–311. [Google Scholar]
- Mocarski, E.S.; Bonyhadi, M.; Salimi, S.; McCune, J.M.; Kaneshima, H. Human cytomegalovirus in a SCID-hu mouse: Thymic epithelial cells are prominent targets of viral replication. Proc. Natl. Acad. Sci. USA 1993, 90, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Kaneshima, H.; Mocarski, E.S. Dramatic Interstrain Differences in the Replication of Human Cytomegalovirus in SCID-hu Mice. J. Infect. Dis. 1995, 171, 1599–1603. [Google Scholar] [CrossRef]
- Smith, M.S.; Goldman, D.C.; Bailey, A.S.; Pfaffle, D.L.; Kreklywich, C.N.; Spencer, D.B.; Othieno, F.A.; Streblow, D.N.; Garcia, J.V.; Fleming, W.H.; et al. Granulocyte-Colony Stimulating Factor Reactivates Human Cytomegalovirus in a Latently Infected Humanized Mouse Model. Cell Host Microbe 2010, 8, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Hakki, M.; Goldman, D.C.; Streblow, D.N.; Hamlin, K.L.; Krekylwich, C.N.; Fleming, W.H.; Nelson, J.A. HCMV Infection of Humanized Mice after Transplantation of G-CSF–Mobilized Peripheral Blood Stem Cells from HCMV-Seropositive Donors. Biol. Blood Marrow Transplant. 2014, 20, 132–135. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Klobuch, S.; Podlech, J.; Plachter, B.; Hoffmann, P.; Renzaho, A.; Theobald, M.; Reddehase, M.; Herr, W.; Lemmermann, N. Evaluating Human T-Cell Therapy of Cytomegalovirus Organ Disease in HLA-Transgenic Mice. PLoS Pathog. 2015, 11, e1005049. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Lemmermann, N.A.W. Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”. Viruses 2018, 10, 693. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J. Mutual Interference between Cytomegalovirus and Reconstitution of Protective Immunity after Hematopoietic Cell Transplantation. Front. Immunol. 2016, 7, 294. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.; Weiland, F.; Münch, K.; Jonjic, S.; Lüske, A.; Koszinowski, U.H. Interstitial murine cytomegalovirus pneumonia after irradiation: Characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 1985, 55, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.; Mutter, W.; Koszinowski, U.H. In vivo application of recombinant interleukin 2 in the immunotherapy of established cytomegalovirus infection. J. Exp. Med. 1987, 165, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.; Podlech, J.; Gillert-Marien, D.; Gergely, K.M.; Büttner, J.K.; Fink, A.; Freitag, K.; Thomas, D.; Reddehase, M.; Holtappels, R. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 527–539. [Google Scholar] [CrossRef]
- Riddell, S.R.; Watanabe, K.; Goodrich, J.; Li, C.; Agha, M.; Greenberg, P. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of Cellular Immunity against Cytomegalovirus in Recipients of Allogeneic Bone Marrow by Transfer of T-Cell Clones from the Donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef]
- Holtappels, R.; Schader, S.I.; Oettel, O.; Podlech, J.; Seckert, C.K.; Reddehase, M.J.; Lemmermann, N.A.W. Insufficient Antigen Presentation Due to Viral Immune Evasion Explains Lethal Cytomegalovirus Organ Disease After Allogeneic Hematopoietic Cell Transplantation. Front. Cell. Infect. Microbiol. 2020, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Gezinir, E.; Podlech, J.; Gergely, K.M.; Becker, S.; Reddehase, M.J.; Lemmermann, N. Enhancement of Antigen Presentation by Deletion of Viral Immune Evasion Genes Prevents Lethal Cytomegalovirus Disease in Minor Histocompatibility Antigen-Mismatched Hematopoietic Cell Transplantation. Front. Cell. Infect. Microbiol. 2020, 10, 279. [Google Scholar] [CrossRef]
- Plotkin, S. The history of vaccination against cytomegalovirus. Med. Microbiol. Immunol. 2015, 204, 247–254. [Google Scholar] [CrossRef]
- Gogesch, P.; Penner, I.; Krauter, S.; Büscher, N.; Grode, L.; Aydin, I.; Plachter, B. Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine. Vaccines 2019, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.A.; Wang, D.; Oualim, A.; Diamond, D.J.; Kotton, C.N.; Mossman, S.; Carfi, A.; Anderson, D.; Dormitzer, P.R. The Status of Vaccine Development Against the Human Cytomegalovirus. J. Infect. Dis. 2020, 221, S113–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Löffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.-G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)–specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mohty, M.; Or, R.; Maschan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Cowan, M.J.; Dunham, K.; Horn, B.; McGuirk, J.; Gilman, A.; Lucas, K.G. Adoptive Immunotherapy With CMV-specific Cytotoxic T Lymphocytes for Stem Cell Transplant Patients With Refractory CMV Infections. J. Immunother. 2012, 35, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odendahl, M.; Grigoleit, G.U.; Bönig, H.; Neuenhahn, M.; Albrecht, J.; Anderl, F.; Germeroth, L.; Schmitz, M.; Bornhäuser, M.; Einsele, H.; et al. Clinical-scale isolation of ‘minimally manipulated’ cytomegalovirus-specific donor lymphocytes for the treatment of refractory cytomegalovirus disease. Cytotherapy 2014, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.-Y.; Zhao, X.-Y.; Chang, Y.-J.; Liu, J.; Xu, L.-P.; Wang, Y.; Zhang, X.-H.; Han, W.; Chen, Y.-H.; Huang, X. Cytomegalovirus-Specific T-Cell Transfer for Refractory Cytomegalovirus Infection After Haploidentical Stem Cell Transplantation: The Quantitative and Qualitative Immune Recovery for Cytomegalovirus. J. Infect. Dis. 2017, 216, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Kaeuferle, T.; Krauss, R.; Blaeschke, F.; Willier, S.; Feuchtinger, T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Chemaly, R.F.; Chou, S.; Einsele, H.; Griffiths, P.; Avery, R.; Razonable, R.R.; Mullane, K.M.; Kotton, C.; Lundgren, J.; Komatsu, T.E.; et al. Definitions of Resistant and Refractory Cytomegalovirus Infection and Disease in Transplant Recipients for Use in Clinical Trials. Clin. Infect. Dis. 2019, 68, 1420–1426. [Google Scholar] [CrossRef]
- McLaughlin-Taylor, E.; Pande, H.; Forman, S.J.; Tanamachi, B.; Li, C.-R.; Zaia, J.A.; Greenberg, P.D.; Riddell, S.R. Identification of the major late human cytomegalovirus matrix protein pp65 as a target antigen for CD8+ virus-specific cytotoxic T lymphocytes. J. Med. Virol. 1994, 43, 103–110. [Google Scholar] [CrossRef]
- Wills, M.; Carmichael, A.J.; Mynard, K.; Jin, X.; Weekes, M.; Plachter, B.; Sissons, J.G. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: Frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol. 1996, 70, 7569–7579. [Google Scholar] [CrossRef] [Green Version]
- Boppana, S.B.; Britt, W.J. Recognition of Human Cytomegalovirus Gene Products by HCMV-Specific Cytotoxic T Cells. Virology 1996, 222, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Weekes, M.; Wills, M.; Mynard, K.; Carmichael, A.J.; Sissons, J.G.P. The Memory Cytotoxic T-Lymphocyte (CTL) Response to Human Cytomegalovirus Infection Contains Individual Peptide-Specific CTL Clones That Have Undergone Extensive Expansion In Vivo. J. Virol. 1999, 73, 2099–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobbold, M.; Khan, N.; Pourgheysari, B.; Tauro, S.; McDonald, D.; Osman, H.; Assenmacher, M.; Billingham, L.; Steward, C.; Crawley, C.; et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA–peptide tetramers. J. Exp. Med. 2005, 202, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.; Tonn, T.; Busch, D.H.; Grigoleit, G.U.; Einsele, H.; Odendahl, M.; Germeroth, L.; Ringhoffer, M.; Ringhoffer, S.; Wiesneth, M.; et al. Adoptive transfer and selective reconstitution of streptamer-selected cytomegalovirus-specific CD8+ T cells leads to virus clearance in patients after allogeneic peripheral blood stem cell transplantation. Transfusion 2010, 51, 591–599. [Google Scholar] [CrossRef]
- Nauerth, M.; Weissbrich, B.; Knall, R.; Franz, T.; Dössinger, G.; Bet, J.; Paszkiewicz, P.J.; Pfeifer, L.; Bunse, M.; Uckert, W.; et al. TCR-Ligand koff Rate Correlates with the Protective Capacity of Antigen-Specific CD8+ T Cells for Adoptive Transfer. Sci. Transl. Med. 2013, 5, 192ra87. [Google Scholar] [CrossRef] [Green Version]
- Schub, A.; Schuster, I.G.; Hammerschmidt, W.; Moosmann, A. CMV-Specific TCR-Transgenic T Cells for Immunotherapy. J. Immunol. 2009, 183, 6819–6830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Klobuch, S.; Besold, K.; Plachter, B.; Dörrie, J.; Schaft, N.; Theobald, M.; Herr, W. Strong and sustained effector function of memory- versus naïve-derived T cells upon T-cell receptor RNA transfer: Implications for cellular therapy. Eur. J. Immunol. 2012, 42, 3442–3453. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtappels, R.; Ebert, S.; Podlech, J.; Fink, A.; Böhm, V.; Lemmermann, N.A.W.; Freitag, K.; Renzaho, A.; Thomas, D.; Reddehase, M.J. Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 352–379. [Google Scholar]
- Munks, M.W.; Gold, M.C.; Zajac, A.L.; Doom, C.M.; Morello, C.; Spector, D.; Hill, A.B. Genome-Wide Analysis Reveals a Highly Diverse CD8 T Cell Response to Murine Cytomegalovirus. J. Immunol. 2006, 176, 3760–3766. [Google Scholar] [CrossRef]
- Holtappels, R.; Simon, C.O.; Munks, M.W.; Thomas, D.; Deegen, P.; Kühnapfel, B.; Däubner, T.; Emde, S.F.; Podlech, J.; Grzimek, N.K.A.; et al. Subdominant CD8 T-Cell Epitopes Account for Protection against Cytomegalovirus Independent of Immunodomination. J. Virol. 2008, 82, 5781–5796. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.; Lemmermann, N.; Thomas, D.; Renzaho, A.; Reddehase, M.J.; Holtappels, R. Immune control in the absence of immunodominant epitopes: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 541–550. [Google Scholar] [CrossRef]
- Holtappels, R.; Lemmermann, N.; Podlech, J.; Ebert, S.; Reddehase, M.J. Reconstitution of CD8 T Cells Protective against Cytomegalovirus in a Mouse Model of Hematopoietic Cell Transplantation: Dynamics and Inessentiality of Epitope Immunodominance. Front. Immunol. 2016, 7, 232. [Google Scholar] [CrossRef]
- Jarvis, M.A.; Hansen, S.G.; Nelson, J.A.; Picker, L.J.; Früh, K. Vaccine vectors using the unique biology and immunology of cytomegalovirus. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 450–562. [Google Scholar]
- Früh, K.; Picker, L. CD8+ T cell programming by cytomegalovirus vectors: Applications in prophylactic and therapeutic vaccination. Curr. Opin. Immunol. 2017, 47, 52–56. [Google Scholar] [CrossRef]
- Caposio, P.; Worm, S.V.D.; Crawford, L.; Perez, W.; Kreklywich, C.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ratts, R.; Marshall, E.E.; et al. Characterization of a live-attenuated HCMV-based vaccine platform. Sci. Rep. 2019, 9, 19236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez, A.C.; Rodríguez-Rojas, C.; Del Val, M. Vaccine vectors: The bright side of cytomegalovirus. Med. Microbiol. Immunol. 2019, 208, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Freitag, K.; Renzaho, A.; Becker, S.; Lemmermann, N.A.; Reddehase, M.J. Revisiting CD8 T-cell ‘Memory Inflation’: New Insights with Implications for Cytomegaloviruses as Vaccine Vectors. Vaccines 2020, 8, 402. [Google Scholar] [CrossRef]
- Wagner, M.; Jonjić, S.; Koszinowski, U.H.; Messerle, M. Systematic Excision of Vector Sequences from the BAC-Cloned Herpesvirus Genome during Virus Reconstitution. J. Virol. 1999, 73, 7056–7060. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, R.; Lemmermann, N.; Thomas, D.; Renzaho, A.; Reddehase, M. Identification of an atypical CD8 T cell epitope encoded by murine cytomegalovirus ORF-M54 gaining dominance after deletion of the immunodominant antiviral CD8 T cell specificities. Med. Microbiol. Immunol. 2015, 204, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Griessl, M.; Renzaho, A.; Freitag, K.; Seckert, C.K.; Reddehase, M.J.; Lemmermann, N.A.W. Stochastic Episodes of Latent Cytomegalovirus Transcription Drive CD8 T-Cell “Memory Inflation” and Avoid Immune Evasion. Front. Immunol. 2021, 12, 668885. [Google Scholar] [CrossRef] [PubMed]
- Böhm, V.; Simon, C.O.; Podlech, J.; Seckert, C.K.; Gendig, D.; Deegen, P.; Gillert-Marien, D.; Lemmermann, N.; Holtappels, R.; Reddehase, M.J. The Immune Evasion Paradox: Immunoevasins of Murine Cytomegalovirus Enhance Priming of CD8 T Cells by Preventing Negative Feedback Regulation. J. Virol. 2008, 82, 11637–11650. [Google Scholar] [CrossRef] [Green Version]
- Seckert, C.K.; Griessl, M.; Büttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kühnapfel, B.; Grzimek, N.K.A.; Reddehase, M.J. Viral latency drives ‘memory inflation’: A unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef]
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef]
- Welten, S.; Sandu, I.; Baumann, N.S.; Oxenius, A. Memory CD8 T cell inflation vs tissue-resident memory T cells: Same patrollers, same controllers? Immunol. Rev. 2018, 283, 161–175. [Google Scholar] [CrossRef]
- Cicin-Sain, L. Cytomegalovirus memory inflation and immune protection. Med. Microbiol. Immunol. 2019, 208, 339–347. [Google Scholar] [CrossRef]
- Welten, S.; Baumann, N.S.; Oxenius, A. Fuel and brake of memory T cell inflation. Med. Microbiol. Immunol. 2019, 208, 329–338. [Google Scholar] [CrossRef]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; van Dommelen, S.; Shellam, G.R.; Hill, A.B. Memory Inflation during Chronic Viral Infection Is Maintained by Continuous Production of Short-Lived, Functional T Cells. Immunity 2008, 29, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, N.S.; Torti, N.; Welten, S.P.M.; Barnstorf, I.; Borsa, M.; Pallmer, K.; Oduro, J.D.; Cicin-Sain, L.; Ikuta, K.; Ludewig, B.; et al. Tissue maintenance of CMV-specific inflationary memory T cells by IL-15. PLoS Pathog. 2018, 14, e1006993. [Google Scholar] [CrossRef]
- Obar, J.; Lefrançois, L. Early events governing memory CD8+ T-cell differentiation. Int. Immunol. 2010, 22, 619–625. [Google Scholar] [CrossRef]
- Smith, C.J.; Venturi, V.; Quigley, M.F.; Turula, H.; Gostick, E.; Ladell, K.; Hill, B.J.; Himelfarb, D.; Quinn, K.M.; Greenaway, H.Y.; et al. Stochastic Expansions Maintain the Clonal Stability of CD8+T Cell Populations Undergoing Memory Inflation Driven by Murine Cytomegalovirus. J. Immunol. 2020, 204, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.E.; Sedikides, G.X.; Okecha, G.; Wills, M.R. Generation, maintenance and tissue distribution of T cell responses to human cytomegalovirus in lytic and latent infection. Med. Microbiol. Immunol. 2019, 208, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Adler, S.P.; Reddehase, M.J. Pediatric roots of cytomegalovirus recurrence and memory inflation in the elderly. Med. Microbiol. Immunol. 2019, 208, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Appay, V.; Koschella, M.; Panther, E.; Roth, E.; Hislop, A.D.; Rickinson, A.B.; Rowland-Jones, S.L.; Blum, H.E.; Pircher, H. Increased Expression of the NK Cell Receptor KLRG1 by Virus-Specific CD8 T Cells during Persistent Antigen Stimulation. J. Virol. 2005, 79, 12112–12116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podlech, J.; Holtappels, R.; Pahl-Seibert, M.-F.; Steffens, H.-P.; Reddehase, M.J. Murine Model of Interstitial Cytomegalovirus Pneumonia in Syngeneic Bone Marrow Transplantation: Persistence of Protective Pulmonary CD8-T-Cell Infiltrates after Clearance of Acute Infection. J. Virol. 2000, 74, 7496–7507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrington, L.A.; Smith, T.A.; Grey, F.; Hill, A.B.; Snyder, C.M. Competition for Antigen at the Level of the APC Is a Major Determinant of Immunodominance during Memory Inflation in Murine Cytomegalovirus Infection. J. Immunol. 2013, 190, 3410–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, N.S.; Welten, S.; Torti, N.; Pallmer, K.; Borsa, M.; Barnstorf, I.; Oduro, J.D.; Cicin-Sain, L.; Oxenius, A. Early primed KLRG1- CMV-specific T cells determine the size of the inflationary T cell pool. PLoS Pathog. 2019, 15, e1007785. [Google Scholar] [CrossRef] [Green Version]
- Grassmann, S.; Mihatsch, L.; Mir, J.; Kazeroonian, A.; Rahimi, R.; Flommersfeld, S.; Schober, K.; Hensel, I.; Leube, J.; Pachmayr, L.O.; et al. Early emergence of T central memory precursors programs clonal dominance during chronic viral infection. Nat. Immunol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Abassi, L.; Cicin-Sain, L. The avid competitors of memory inflation. Curr. Opin. Virol. 2020, 44, 162–168. [Google Scholar] [CrossRef]
- Dorsch-Häsler, K.; Keil, G.M.; Weber, F.; Jasin, M.; Schaffner, W.; Koszinowski, U.H. A long and complex enhancer activates transcription of the gene coding for the highly abundant immediate early mRNA in murine cytomegalovirus. Proc. Natl. Acad. Sci. USA 1985, 82, 8325–8329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmermann, N.A.W.; Kropp, K.A.; Seckert, C.K.; Grzimek, N.K.A.; Reddehase, M.J. Reverse Genetics Modification of Cytomegalovirus Antigenicity and Immunogenicity by CD8 T-Cell Epitope Deletion and Insertion. J. Biomed. Biotechnol. 2010, 2011, 812742. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitag, K.; Hamdan, S.; Reddehase, M.J.; Holtappels, R. Immunodominant Cytomegalovirus Epitopes Suppress Subdominant Epitopes in the Generation of High-Avidity CD8 T Cells. Pathogens 2021, 10, 956. https://doi.org/10.3390/pathogens10080956
Freitag K, Hamdan S, Reddehase MJ, Holtappels R. Immunodominant Cytomegalovirus Epitopes Suppress Subdominant Epitopes in the Generation of High-Avidity CD8 T Cells. Pathogens. 2021; 10(8):956. https://doi.org/10.3390/pathogens10080956
Chicago/Turabian StyleFreitag, Kirsten, Sara Hamdan, Matthias J. Reddehase, and Rafaela Holtappels. 2021. "Immunodominant Cytomegalovirus Epitopes Suppress Subdominant Epitopes in the Generation of High-Avidity CD8 T Cells" Pathogens 10, no. 8: 956. https://doi.org/10.3390/pathogens10080956
APA StyleFreitag, K., Hamdan, S., Reddehase, M. J., & Holtappels, R. (2021). Immunodominant Cytomegalovirus Epitopes Suppress Subdominant Epitopes in the Generation of High-Avidity CD8 T Cells. Pathogens, 10(8), 956. https://doi.org/10.3390/pathogens10080956