
Puumala Virus Variants Circulating in Forests of Ardennes, France: Ten Years of Genetic Evolution

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
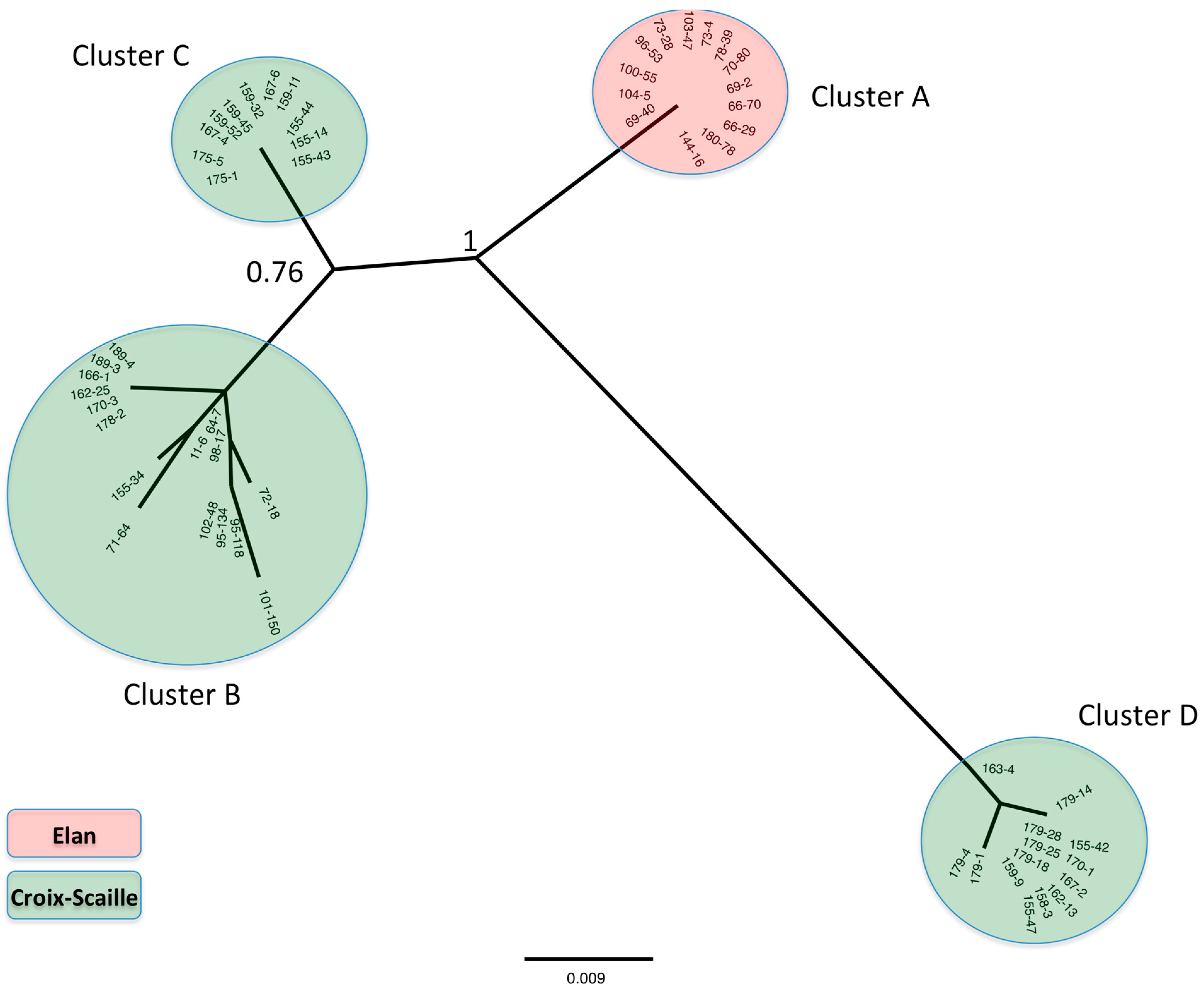
2.1. PUUV Microevolution
2.1.1. Phylogenetic Analysis
2.1.2. Genetic Diversity of PUUV Isolates
2.1.3. Evolution of Genetic Diversity over Time
2.2. Population Dynamics of Rodents
3. Discussion
4. Materials and Methods
4.1. Rodent Trapping and Collection Data
4.2. Serological and Molecular Analysis
4.3. PUUV Microevolution
4.4. Bank Vole Population Dynamics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plyusnin, A.; Sironen, T. Evolution of Hantaviruses: Co-Speciation with Reservoir Hosts for More than 100 MYR. Virus Res. 2014, 187, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus Infections in Europe and Their Impact on Public Health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef]
- Guivier, E.; Galan, M.; Malé, P.-J.G.; Kallio, E.R.; Voutilainen, L.; Henttonen, H.; Olsson, G.E.; Lundkvist, A.; Tersago, K.; Augot, D.; et al. Associations between MHC Genes and Puumala virus Infection in Myodes glareolus Are Detected in Wild Populations, but Not from Experimental Infection Data. J. Gen. Virol. 2010, 91, 2507–2512. [Google Scholar] [CrossRef]
- Deter, J.; Bryja, J.; Chaval, Y.; Galan, M.; Henttonen, H.; Laakkonen, J.; Voutilainen, L.; Vapalahti, O.; Vaheri, A.; Salvador, A.R.; et al. Association between the DQA MHC Class II Gene and Puumala virus Infection in Myodes glareolus, the Bank Vole. Infect. Genet. Evol. 2008, 8, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Rang, A.; Lütteke, N.; Raftery, M.J.; Charbonnel, N.; Ulrich, R.G. Hantavirus-Induced Immunity in Rodent Reservoirs and Humans. Immunol. Rev. 2008, 225, 163–189. [Google Scholar] [CrossRef]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the Mysteries of Hantavirus Infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Olsson, G.E.; Leirs, H.; Henttonen, H. Hantaviruses and Their Hosts in Europe: Reservoirs Here and There, but Not Everywhere? Vector Borne Zoonotic Dis. 2010, 10, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Augot, D.; Sauvage, F.; Boue, F.; Bouloy, M.; Artois, M.; Demerson, J.M.; Combes, B.; Coudrier, D.; Zeller, H.; Cliquet, F.; et al. Spatial and Temporal Patterning of Bank Vole Demography and the Epidemiology of the Puumala hantavirus in Northeastern France. Epidemiol. Infect. 2008, 136, 1638–1643. [Google Scholar] [CrossRef]
- Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.-B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.-F.; Plyusnin, A.; Marianneau, P.; et al. Complete Genome and Phylogeny of Puumala hantavirus Isolates Circulating in France. Viruses 2015, 7, 5476–5488. [Google Scholar] [CrossRef] [Green Version]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular Evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faber, M.; Krüger, D.H.; Auste, B.; Stark, K.; Hofmann, J.; Weiss, S. Molecular and Epidemiological Characteristics of Human Puumala and Dobrava-Belgrade Hantavirus Infections, Germany, 2001 to 2017. Eurosurveillance 2019, 24, 1800675. [Google Scholar] [CrossRef]
- Plyusnin, A.; Vapalahti, O.; Lehväslaiho, H.; Apekina, N.; Mikhailova, T.; Gavrilovskaya, I.; Laakkonen, J.; Niemimaa, J.; Henttonen, H.; Brummer-Korvenkontio, M. Genetic Variation of Wild Puumala virus within the Serotype, Local Rodent Populations and Individual Animal. Virus Res. 1995, 38, 25–41. [Google Scholar] [CrossRef]
- Lundkvist, A.; Wiger, D.; Hörling, J.; Sjölander, K.B.; Plyusnina, A.; Mehl, R.; Vaheri, A.; Plyusnin, A. Isolation and Characterization of Puumala hantavirus from Norway: Evidence for a Distinct Phylogenetic Sublineage. J. Gen. Virol. 1998, 79 Pt 11, 2603–2614. [Google Scholar] [CrossRef]
- Rowe, J.E.; St Jeor, S.C.; Riolo, J.; Otteson, E.W.; Monroe, M.C.; Henderson, W.W.; Ksiazek, T.G.; Rollin, P.E.; Nichol, S.T. Coexistence of Several Novel Hantaviruses in Rodents Indigenous to North America. Virology 1995, 213, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of Point Mutations and Reassortment of Genomic RNA Segments Are Involved in the Microevolution of Puumala hantavirus in a Bank Vole (Myodes glareolus) Population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a Bank Vole Population in Northern Finland: Evidence for Co-Circulation of Two Genetic Lineages and Frequent Reassortment between Strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicker, D.G.; Lemey, P.; Velasco-Villa, A.; Rupprecht, C.E. Rates of Viral Evolution Are Linked to Host Geography in Bat Rabies. PLoS Pathog. 2012, 8, e1002720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala hantavirus during a Complete Population Cycle of Its Host, the Bank Vole (Myodes glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [Green Version]
- Escutenaire, S.; Chalon, P.; Verhagen, R.; Heyman, P.; Thomas, I.; Karelle-Bui, L.; Avsic-Zupanc, T.; Lundkvist, A.; Plyusnin, A.; Pastoret, P. Spatial and Temporal Dynamics of Puumala hantavirus Infection in Red Bank Vole (Clethrionomys glareolus) Populations in Belgium. Virus Res. 2000, 67, 91–107. [Google Scholar] [CrossRef]
- Olsson, G.E.; White, N.; Ahlm, C.; Elgh, F.; Verlemyr, A.-C.; Juto, P.; Palo, R.T. Demographic Factors Associated with Hantavirus Infection in Bank Voles (Clethrionomys glareolus). Emerg. Infect. Dis. 2002, 8, 924–929. [Google Scholar] [CrossRef]
- Mazurkiewicz, M.; Rajska-Jurgiel, E. Spatial Behaviour and Population Dynamics of Woodland Rodents. Acta Theriol. 1998, 43, 137–161. [Google Scholar] [CrossRef] [Green Version]
- Klempa, B. Hantaviruses and Climate Change. Clin. Microbiol. Infect. 2009, 15, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Prist, P.R.; Uriarte, M.; Fernandes, K.; Metzger, J.P. Climate Change and Sugarcane Expansion Increase Hantavirus Infection Risk. PLoS Negl. Trop. Dis. 2017, 11, e0005705. [Google Scholar] [CrossRef] [Green Version]
- Serra-Cobo, J. Emerging Infectious Diseases in a Globalized World. Acad. J. Health Sci. 2021, 38–41. [Google Scholar] [CrossRef]
- Voutilainen, L.; Sironen, T.; Tonteri, E.; Bäck, A.T.; Razzauti, M.; Karlsson, M.; Wahlström, M.; Niemimaa, J.; Henttonen, H.; Lundkvist, Å. Life-Long Shedding of Puumala hantavirus in Wild Bank Voles (Myodes glareolus). J. Gen. Virol. 2015, 96, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, F.; Penalba, C.; Vuillaume, P.; Boue, F.; Coudrier, D.; Pontier, D.; Artois, M. Puumala hantavirus Infection in Humans and in the Reservoir Host, Ardennes Region, France. Emerg. Infect. Dis. 2002, 8, 1509–1511. [Google Scholar] [CrossRef]
- Heyman, P.; Cochez, C.; Ducoffre, G.; Mailles, A.; Zeller, H.; Abu Sin, M.; Koch, J.; van Doornum, G.; Koopmans, M.; Mossong, J.; et al. Haemorrhagic Fever with Renal Syndrome: An Analysis of the Outbreaks in Belgium, France, Germany, the Netherlands and Luxembourg in 2005. Eurosurveillance 2007, 12, E15–E16. [Google Scholar] [CrossRef]
- Reynes, J.-M.; Matheus, S.; Lavergne, A. Rapport Annuel D’activité Du Centre National de Référence Des Hantavirus, Année D’exercice 2018–2019. Available online: https://www.pasteur.fr/fr/file/26905/download (accessed on 27 August 2021).
- Reynes, J.-M.; Carli, D.; Renaudin, B.; Fizet, A.; Bour, J.-B.; Brodard, V.; Cart-Tanneur, E.; Dewilde, A.; El Hamri, M.; Fleury, H.; et al. Surveillance Des Infections Humaines Par Hantavirus En France Métropolitaine, 2012–2016. Bull. Epidémiol. Hebd. 2017, 23, 492–499. [Google Scholar]
- Kendall, W.L.; Pollock, K.H.; Brownie, C. A Likelihood-Based Approach to Capture-Recapture Estimation of Demographic Parameters under the Robust Design. Biometrics 1995, 51, 293. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Severson, W.; Villegas, N.; Schmaljohn, C.S.; Jonsson, C.B. The RNA Binding Domain of the Hantaan Virus N Protein Maps to a Central, Conserved Region. J. Virol. 2002, 76, 3301–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaukinen, P.; Vaheri, A.; Plyusnin, A. Hantavirus Nucleocapsid Protein: A Multifunctional Molecule with Both Housekeeping and Ambassadorial Duties. Arch. Virol. 2005, 150, 1693–1713. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Vrancken, B.; Maes, P. Identifying the Patterns and Drivers of Puumala hantavirus Enzootic Dynamics Using Reservoir Sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef] [Green Version]
- Guivier, E.; Galan, M.; Chaval, Y.; Xuéreb, A.; Ribas Salvador, A.; Poulle, M.-L.; Voutilainen, L.; Henttonen, H.; Charbonnel, N.; Cosson, J.F. Landscape Genetics Highlights the Role of Bank Vole Metapopulation Dynamics in the Epidemiology of Puumala hantavirus. Mol. Ecol. 2011, 20, 3569–3583. [Google Scholar] [CrossRef] [PubMed]
- Quéré, J.-P.; le Louarn, H.; ProQuest (Firme). Les rongeurs de France Faunistique et Biologie; Éditions Quæ: Versailles, France, 2011. [Google Scholar]
- Kallio, E.R.; Klingström, J.; Gustafsson, E.; Manni, T.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; Lundkvist, Å. Prolonged Survival of Puumala hantavirus Outside the Host: Evidence for Indirect Transmission via the Environment. J. Gen. Virol. 2006, 87, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Ecke, F.; Evander, M.; Bucht, G.; Hörnfeldt, B. Population Dynamics of Bank Voles Predicts Human Puumala Hantavirus Risk. Ecohealth 2019, 16, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Forbes, K.M.; Mappes, T.; Sironen, T.; Strandin, T.; Stuart, P.; Meri, S.; Vapalahti, O.; Henttonen, H.; Huitu, O. Food Limitation Constrains Host Immune Responses to Nematode Infections. Biol. Lett. 2016, 12, 20160471. [Google Scholar] [CrossRef] [Green Version]
- Kusumoto, K. Effect of Food Availability on Immune Function of Gray Red-Backed Voles (Myodes rufocanus) Exposed to Low Temperature and Short Photoperiod. J. Mammal. 2009, 90, 416–422. [Google Scholar] [CrossRef]
- Lundkvist, A.; Cheng, Y.; Sjölander, K.B.; Niklasson, B.; Vaheri, A.; Plyusnin, A. Cell Culture Adaptation of Puumala hantavirus Changes the Infectivity for Its Natural Reservoir, Clethrionomys glareolus, and Leads to Accumulation of Mutants with Altered Genomic RNA S Segment. J. Virol. 1997, 71, 9515–9523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sironen, T.; Kallio, E.R.; Vaheri, A.; Lundkvist, A.; Plyusnin, A. Quasispecies Dynamics and Fixation of a Synonymous Mutation in Hantavirus Transmission. J. Gen. Virol. 2008, 89, 1309–1313. [Google Scholar] [CrossRef]
- Heyman, P.; Ceianu, C.S.; Christova, I.; Tordo, N.; Beersma, M.; João Alves, M.; Lundkvist, A.; Hukic, M.; Papa, A.; Tenorio, A.; et al. A Five-Year Perspective on the Situation of Haemorrhagic Fever with Renal Syndrome and Status of the Hantavirus Reservoirs in Europe, 2005–2010. Eurosurveillance 2011, 16, 19961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynes, J.-M.; Dutrop, C.-M.; Carli, D.; Levast, M.; Fontaine, N.; Denoyel, G.-A.; Philit, J.-B. Puumala hantavirus infection in Isère: Geographic extension of this zoonosis in France. Med. Mal. Infect. 2015, 45, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Klempa, B.; Tenner, B.; Kruger, D.H.; Hofmann, J. Prediction of the Spatial Origin of Puumala virus Infections Using L Segment Sequences Derived from a Generic Screening PCR. Viruses 2019, 11, 694. [Google Scholar] [CrossRef] [Green Version]
- Plyusnina, A.; Razzauti, M.; Sironen, T.; Niemimaa, J.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Plyusnin, A. Analysis of Complete Puumala virus Genome, Finland. Emerg. Infect. Dis. 2012, 18, 2070–2072. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, G.C.; Burnham, K.P. Program MARK: Survival Estimation from Populations of Marked Animals. Bird Study 1999, 46, S120–S139. [Google Scholar] [CrossRef]
- Cormack, R.M. Estimates of Survival from the Sighting of Marked Animals. Biometrika 1964, 51, 429–438. [Google Scholar] [CrossRef]
- Jolly, G.M. Explicit Estimates from Capture-Recapture Data with Both Death and Immigration-Stochastic Model. Biometrika 1965, 52, 225–248. [Google Scholar] [CrossRef]
- Seber, G.A.F. A Note on the Multiple-Recapture Census. Biometrika 1965, 52, 249–260. [Google Scholar] [CrossRef]
- Kendall, W.L.; Bjorkland, R. Using Open Robust Design Models to Estimate Temporary Emigration from Capture-Recapture Data. Biometrics 2001, 57, 1113–1122. [Google Scholar] [CrossRef]
- Pollock, K.H. A Capture-Recapture Design Robust to Unequal Probability of Capture. J. Wildl. Manag. 1982, 46, 752. [Google Scholar] [CrossRef]
- Bailey, L.L.; Simons, T.R.; Pollock, K.H. Estimating Detection Probability Parameters for Plethodon Salamanders Using the Robust Capture-Recapture Design. J. Wildl. Manag. 2004, 68, 1–13. [Google Scholar] [CrossRef]
- Burnham, K.P.; Anderson, D.R. Model. Selection and Multimodel Inference: A Practical Information-Theoretic Approach, 2nd ed.; Springer: New York, NY, USA, 2010; ISBN 978-0-387-95364-9. [Google Scholar]
- Sauer, J.R.; Williams, B.K. Generalized Procedures for Testing Hypotheses about Survival or Recovery Rates. J. Wildl. Manag. 1989, 53, 137. [Google Scholar] [CrossRef]
- Hines, J.E.; Sauer, J.R. Program CONTRAST: A General Program for the Analysis of Several Survival or Recovery Rate Estimates; US Department of the Interior, Fish and Wildlife Service: Washington, DC, USA, 1989. [Google Scholar]
- Pradel, R. Utilization of Capture-Mark-Recapture for the Study of Recruitment and Population Growth Rate. Biometrics 1996, 52, 703. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Station 2 | Station 3 | Station 4 | Station 5 | Elan Forest | Croix-Scaille Forest | |
|---|---|---|---|---|---|---|
| Station 2 | 10.75 (3.5) | 12.67 (4.2) | 12.67 (4.2) | 10.62 (3.5) | ND | ND |
| Station 3 | 0 (0) | 0 (0) | 12.64 (4.2) | ND | ND | |
| Station 4 | 0 (0) | 12.64 (4.2) | ND | ND | ||
| Station 5 | 8.51 (2.8) | ND | ND | |||
| Elan forest | 0 (0) | ND | ||||
| Croix-Scaille forest | 19.3 (3.2) |
| Station (Forest) | Detection Period | Diversification (Number of Different Variants for Each Cluster) | Total Number of Different Variants for Each Station | |||
|---|---|---|---|---|---|---|
| Cluster A | Cluster B | Cluster C | Cluster D | |||
| 3 (Elan) | 2003–2009 | 1 | NO | NO | NO | 1 |
| 4 (Elan) | 2003–2005 | 1 | NO | NO | NO | 1 |
| 2 (Croix-Scaille) | 2000–2009 | NO | 5 | 1 | 4 | 10 |
| 5 (Croix-Scaille) | 2003–2009 | NO | 3 | 0 | 1 | 4 |
| Station | Survival (ϕ) a | Seniority (Υ) a | Recruitment (f) b | Population Growth (λ) b |
|---|---|---|---|---|
| Croix-Scaille | ||||
| St5 | 0.094 (0.015; 0.421) | 0.071 (0.012; 0.329) | 1.230 (0.839; 1.264) | 1.324 (0.854; 1.685) |
| St2 | 0.231 (0.115; 0.410) | 0.186 (0.100; 0.319]) | 1.011 (0.783; 1.157) | 1.242 (0.898; 1.567) |
| Elan | ||||
| St3 | 0.160 (0.077; 0.303) | 0.134 (0.070; 0.241]) | 1.034 (0.835; 1.169) | 1.194 (0.912; 1.472) |
| St4 | 0.315 (0.183; 0.485) | 0.267 (0.167; 0.399]) | 0.865 (0.659; 1.013) | 1.180 (0.842; 1.498) |
| PUUV Diversity | Population Dynamics | Seroprevalence (SP) | Environment | |
|---|---|---|---|---|
| Elan Forest | ||||
| Station 3 | No genetic diversity over time | Less extinction over time, peaks some years, good survival | Lower SP than in Croix-Scaille forest with peak in 2003 | A limited forest of broadleaves, surrounded by fields and roads |
| Station 4 | No genetic diversity over time | Less extinction over time, peaks some years, good survival | Lower SP than at other stations | |
| Croix-Scaille Forest | ||||
| Station 2 | Highest genetic diversity over time and at each time | Overall good survival and abundance similar to Elan forest but extinction some years | Very high SP every year bank voles are present | Oaks with conifer plots in a large forest massif with harsher weather conditions than in Elan |
| Station 5 | Lower genetic diversity than station 2 and higher than Elan forest | Numerous extinctions, lowest survival, and low abundance | Highest SP every year bank voles are present | Coniferous plot in a large forest massif with harsher weather conditions than in Elan |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castel, G.; Monchatre-Leroy, E.; López-Roig, M.; Murri, S.; Couteaudier, M.; Boué, F.; Augot, D.; Sauvage, F.; Pontier, D.; Hénaux, V.; et al. Puumala Virus Variants Circulating in Forests of Ardennes, France: Ten Years of Genetic Evolution. Pathogens 2021, 10, 1164. https://doi.org/10.3390/pathogens10091164
Castel G, Monchatre-Leroy E, López-Roig M, Murri S, Couteaudier M, Boué F, Augot D, Sauvage F, Pontier D, Hénaux V, et al. Puumala Virus Variants Circulating in Forests of Ardennes, France: Ten Years of Genetic Evolution. Pathogens. 2021; 10(9):1164. https://doi.org/10.3390/pathogens10091164
Chicago/Turabian StyleCastel, Guillaume, Elodie Monchatre-Leroy, Marc López-Roig, Séverine Murri, Mathilde Couteaudier, Franck Boué, Denis Augot, Frank Sauvage, Dominique Pontier, Viviane Hénaux, and et al. 2021. "Puumala Virus Variants Circulating in Forests of Ardennes, France: Ten Years of Genetic Evolution" Pathogens 10, no. 9: 1164. https://doi.org/10.3390/pathogens10091164
APA StyleCastel, G., Monchatre-Leroy, E., López-Roig, M., Murri, S., Couteaudier, M., Boué, F., Augot, D., Sauvage, F., Pontier, D., Hénaux, V., Marianneau, P., Serra-Cobo, J., & Tordo, N. (2021). Puumala Virus Variants Circulating in Forests of Ardennes, France: Ten Years of Genetic Evolution. Pathogens, 10(9), 1164. https://doi.org/10.3390/pathogens10091164