1. Introduction
Filarioid nematodes are a group of tissue-dwelling nematodes of vertebrates, transmitted by hematophagous arthropods.
Stephanofilaria spp. are small (up to 8 mm) parasitic roundworms, causing cutaneous lesions mainly in cattle, but have also been reported to cause dermatitis in other livestock and mammalian wildlife species including buffaloes, goats, pigs, rhinoceroses, hippopotami, and giraffes [
1,
2,
3,
4,
5,
6,
7]. Although the systematics of this group is somewhat fluid, a recent placement of the genus
Stephanofilaria in the Nematoda is order Spirurida, superfamily Filariodidea, family Filarioidea [
8].
Stephanofilaria stilesi,
S. assamensis,
S. dedoesi,
S. kaeli and
S. okinawaensis have been reported in cattle skin lesions from the United States, Russia, India, Indonesia, Malaysia, and Japan, respectively (reviewed by Johnson [
6]).
In Australia, cattle are infested by an unnamed species of
Stephanofilaria. This species is found widely in cattle in northern Australia in association with the buffalo fly,
Haematobia irritans exigua (BF). Johnson [
6] has provided a full description of the Australian
Stephanofilaria sp. complete with detailed morphological diagrams, electron microscope images, and a tentative key to species, including the Australian type. He differentiated the Australian
Stephanofilaria from
S. stilesi on the basis of the cephalic armature, with 4–6 cephalic spines present in
S. stilesi but absent in the Australian type. However, he did not elevate the Australian type to species status because of difficulty in obtaining samples of
S. stilesi for detailed comparison.
In cattle, adult nematodes live in the dermis forming cysts at the base of hair follicles and release membrane-encapsulated microfilariae into the dermal tissues. Microfilariae of
S. stilesi and
Stephanofilaria sp. in Australia are surrounded by a vitelline membrane (up to 60 μm diameter) and are thought to be the stage ingested by vector flies [
6,
7]. The microfilaria develops through the first and second larval stages (L1 and L2) to infective larva (L3) (2–3 weeks) in the intermediate host fly, before being released back into cattle skin during fly feeding [
6,
7]. Further information on the morphology of intermediate stages and epidemiology and pathogenesis of lesions is given by Johnson et al. [
9], Johnson and Toleman [
10], Shaw and Sutherland [
11], and Sutherst et al. [
12], respectively. Horn flies
Haematobia irritans irritans (HF), BF and
Musca conducens have been reported as the main biological vectors for different species of the genus
Stephanofilaria.
In Australia,
Stephanofilaria sp. is transmitted by BF; therefore, lesions are recognised as BF lesions [
9,
13], which are clinically manifested as dermatitis or wounds on the medial canthus of the eye, neck, dewlap, and ventral midline of cattle. This differs from
S. stilesi in which lesions are found on the abdominal midline, sometimes extending to the udder [
14] in cows and scrotum of bulls [
15]. The lesions appear as areas with serous exudation, scab encrustation, ulceration, or circumscribed papules of 1–10 cm in diameter. Chronicity of the lesions leads to alopecia, acanthosis, and hyperkeratosis [
6,
9]. Development of lesions results in permanent hide damage decreasing the value of the hide and may reduce the market value of the affected stock [
12]. Moreover, the presence of lesions is also a significant welfare issue and can increase the susceptibility of cattle to other infections.
Direct visualisation of nematodes in skin sections by microscopic examination or the use of a saline recovery technique [
6] are the only currently available methods for the detection of
Stephanofilaria spp. in skin lesions. A comparison of techniques to diagnose
Stephanofilaria sp. found 12% of lesion biopsies positive for filarial nematodes by the saline recovery technique and 32% by histopathological examination of tissue sections, whereas 10% of the samples that yielded nematodes with the saline recovery technique were found negative by histology [
13]. In contrast, Miyakawa et al. [
14] observed no adult
S. stilesi or microfilariae in histopathological sections of skin lesions, but the same specimens were found to be 100% positive for nematodes using the saline recovery technique. Similarly, detection of
Stephanofilaria sp. in the BF vector is difficult. It involves the dissection of freshly collected flies and the examination of internal contents using microscopy [
6,
11]. These methods are time consuming, laborious, and require training and microscopic expertise. Detection of microfilaria is particularly difficult due to their small size. Currently available diagnostic techniques for
Stephanofilaria sp. in skin lesions and BFs are less sensitive and are laborious and imprecise. Therefore, an accurate, sensitive, and rapid diagnostic method was required.
Many studies have confirmed that PCR-based diagnostic approaches using specific primers can provide sensitive detection and identification of nematodes and their developmental stages [
16,
17]. Assays based on the amplification of internal transcribed spacer regions (ITS) of rDNA have been particularly useful for the detection of nematodes because there is relatively low variation in the ITS sequence within a species but large differences between species, as reported in other members of the Strongylida [
18]. Several studies of PCR-based detection of helminths have been undertaken for the orders Strongylida and Ascaridida [
18,
19], and methods have been published for nematode species in the order Spirurida including
Dirofilaria and
Dipetalonema [
20],
Gnathostoma [
21],
Onchocerca and
Mansonella [
16],
Thelazia [
22],
Habronema [
23], and
Parafilaria [
24].
The sensitivity of molecular techniques for detecting a small quantity of nematode DNA, regardless of the developmental stage, suggested that similar approaches would be effective for the detection of Stephanofilaria sp. Furthermore, it is necessary to accurately detect and identify Stephanofilaria sp., to study its systematics, distribution, and epidemiology, and also to reliably determine the aetiology of skin lesions to design better treatment and control methods. However, no reports using PCR-based diagnosis for any species from the genus Stephanofilaria have been previously published, and no genome sequence information is currently available for this genus in sequence databases. In this study, we determined the cox1 and ITS2 sequences of Stephanofilaria sp. and constructed a phylogeny of the Stephanofilaria sp. cox1 sequence with other closely related nematode genera. In addition, molecular diagnostic assays were developed and optimised using an ITS2 based conventional PCR and qPCR methods for Stephanofilaria sp. detection in cattle skin lesions and the BF vector, respectively.
2. Material and Methods
2.1. Sample Collection
Cattle skin biopsies were collected from an abattoir in Townsville (JBS Australia, Stuart, Queensland, 4811, −19.329863, 146.855438) during two different visits. All biopsies were collected from hides of freshly slaughtered cattle with obvious lesions near the medial canthus of the eyes. Grossly, lesions sampled were raised, alopecic areas of various sizes covered with dry serous crust or scab. In May 2019, biopsies were collected from 10 hides (Labelled as C-1 to C-10), and in September 2019, biopsies were collected from 15 hides (Labelled as C-11 to C-25). All biopsies were collected using 8 mm skin punches (Paramount Surgimed Ltd., New Delhi, India). From each hide, two biopsies were collected, one in normal saline solution and one in absolute ethanol for examination by the saline recovery technique and DNA extraction, respectively.
2.2. Isolation of Nematode and Microfilariae
Each saline solution biopsy was cut into 5–7 slices (1–2 mm thick) using a scalpel blade (size 24, Livingstone, India) on the day of sample collection. The sliced samples were transferred to Petri plates with saline solution (0.9% sodium chloride) and incubated at room temperature overnight (method modified from Johnson [
6]). Each Petri plate was inspected for nematode recovery by two observers under a stereomicroscope (SZX 10, Olympus, Shinjiku, Japan) at 2.4X magnification.
After recovering adult nematodes, the saline solution from each Petri dish was transferred to a 15 mL screw-capped centrifugation tube using disposable plastic transfer pipettes. These tubes were centrifuged (Eppendorf 5702P) at 1500× g for 5 min at 20 °C. The supernatant was discarded, and the sediments were vortexed (VM1 Ratek, VIC, Australia) for 10 s. One drop from the sediments was placed onto a glass slide and covered with a glass coverslip before microscopy at 10X (Axioskop 40, Zeiss, Jena, Germany) for identification of microfilariae. Sediments found positive for microfilariae were preserved in absolute ethanol at −20 °C.
2.3. Morphological Examination by Scanning Electron Microscopy (SEM)
Adult nematodes (n = 10) isolated by saline recovery were morphologically identified using scanning electron microscopy (SEM) at the Centre for Microscopy and Microanalysis, The University of Queensland. Specimens were fixed with 4% osmium, followed by two washes with absolute ethanol to remove the osmium. Thereafter, they were dehydrated by ascending grades (25%, 50%, 75%, and 100%) of hexamethyldisilazane. After critical drying, nematodes were mounted on a carbon stub, coated with gold, and observed under the SEM (Hitachi SU3500, Osaki, Japan). All the SEM procedures were undertaken at The University of Queensland, Centre for Microscopy and Microanalysis facility.
2.4. Nematode DNA Extraction
Two adult nematodes were washed twice with 500 μL of Milli-Q® water in a 1.5 mL tube to remove preserving ethanol. DNA was extracted using a Qiagen DNeasy Blood and Tissue Extraction Kit as described by the manufacturer (QIAGEN Pty Ltd., Hilden, Germany, cat. Nos. 69504 & 69506). Briefly, the worms were homogenised in lysis buffer by 4 freeze cycles (45 s each) in liquid N2 and thawed at 50 °C using a water bath, followed by lysis in a tissue lyser (TissueLyser II, QIAGEN Pty Ltd., Hilden, Germany) with 0.5 mm zirconia beads for 30 Hz/second for 2 min. The remainder of the extraction protocol was as recommended by the manufacturer, with the exception that all extractions were eluted in 30 μL elution buffer (AE buffer). DNA concentration was measured using a NanoDrop spectrophotometer (NanoDrop 2000, Thermofisher Scientific Pty Ltd., Waltham, MA, USA).
2.5. Primer Design, cox1 Amplification, and Sequencing
To amplify the
cox1 gene of mtDNA of
Stephanofilaria sp., mtDNA sequences for
Onchocerca volvulus (accession no. AP017695),
Loa loa (accession no. NC016199)
, Dirofilaria immitis (accession no. AJ537512)
, and
Parafilaria bovicola (accession no. MG983751) were aligned using Geneious software (version 11.1.4, Biomatters Ltd., Auckland, New Zealand). Primers were designed from conserved sequence regions targeting the desired mtDNA product. The
cox1 region of mtDNA was amplified using St_Co1_F and St_Co1_R primers. Primer pair details are described in
Table 1.
PCR was performed in a total volume of 10 μL containing 1 μL of 10X Taq buffer, 200 μM of each dNTP, 1.5 mM of MgCl2, 5 μM of each primer, 0.5 U of Taq DNA polymerase (QIAGEN Pty Ltd., Hilden, Germany, cat. no. 201203) and 3 ng of DNA template. The cycling conditions included an initial denaturation at 95 °C for 2 min, initial primer annealing at 50 °C for 45 s and Taq polymerase activation at 72 °C for 90 s, followed by 30 cycles of denaturation, annealing, and extension at 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 90 s, respectively, with a final extension at 72 °C for 7 min. The amplification reaction was conducted in a T100TM thermal cycler (Bio-Rad, CALIF, Hercules, CA, USA). The PCR products were run on 1% agarose gels containing gel red dye (Biotium, Fremont, CA, USA, cat. no. 41003) with 1X TBE buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA pH 8) for 40 min at 110V using a 1 kb DNA gene ruler ladder (Thermofisher Scientific, Waltham, MA, USA, cat. no. SM0311). Distinct bands were dissected under UV light, and the PCR products were purified using a PureLinkTM Quick gel extraction kit (QIAGEN Pty Ltd., Hilden, Germany, cat. no. K210012). Purified PCR products were sequenced through the Australian Genomic Research Facility (AGRF), The University of Queensland (St. Lucia, 4072, Queensland, Australia).
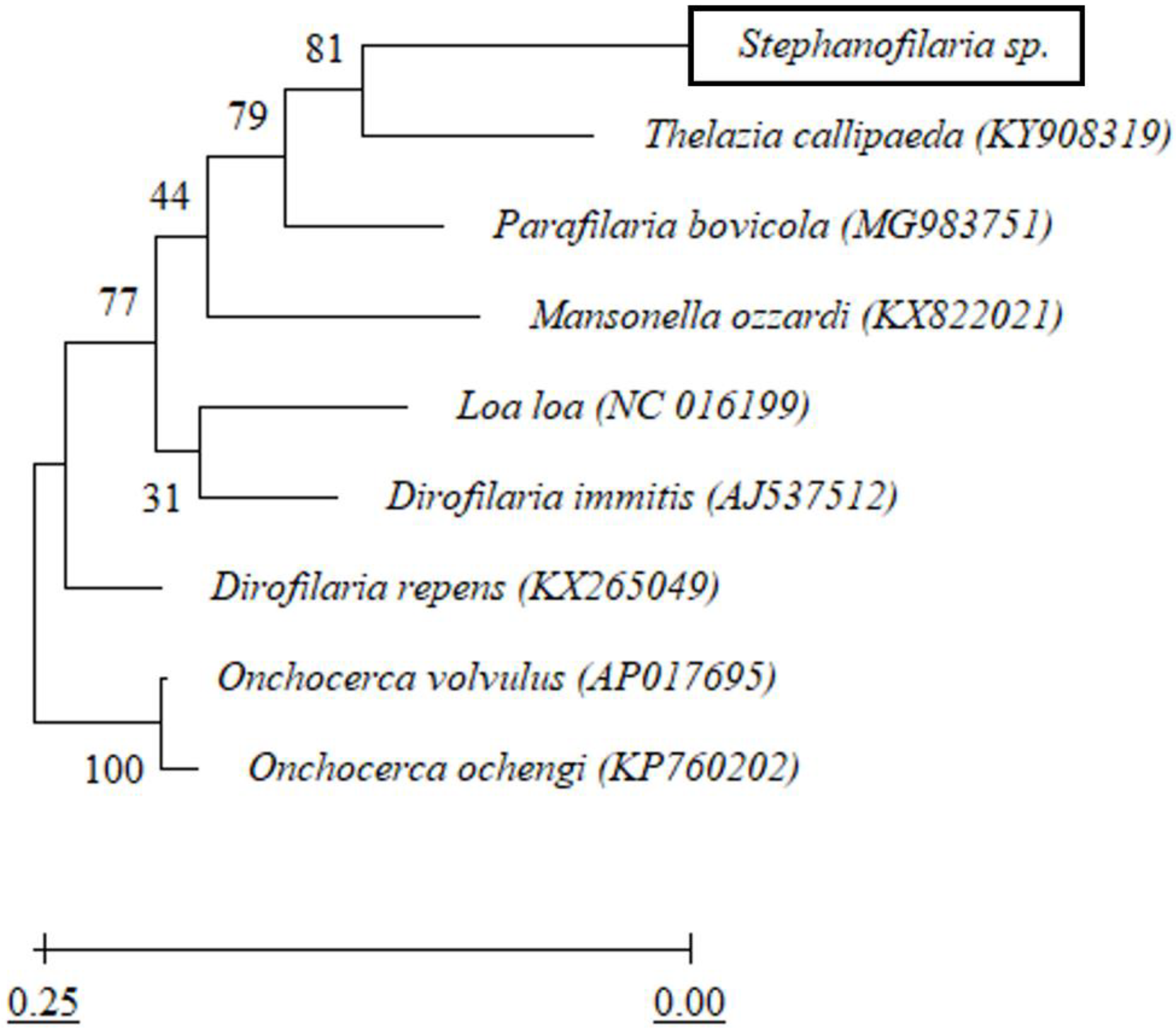
2.6. cox1 Based Phylogenetic Analysis
All forward and reverse sequences for
cox1 were aligned using Geneious software (v11.1.4). Endreads and other obvious errors were corrected following inspection of the DNA sequence electropherograms. The trimmed and edited DNA sequences were used to detect similarities with other available sequences in GenBank using BLASTn (
https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 26 October 2020)). A phylogenetic tree based on
cox1 sequences was constructed using maximum likelihood analysis and a Tamura-Nei model of evolution [
25] in Mega X [
26]. Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-Nei model and then selecting the topology with a superior log-likelihood value. The analysis involved
cox1 nucleotide sequences from eight other closely related filarial nematodes (
Table 2). There was a total of 433 nucleotide positions in the final dataset without gaps. The tree was rooted using
Onchocerca spp. as an outgroup. The robustness of the topology was tested with 1000 bootstraps [
27] using the same program.
2.7. Development and Optimisation of Stephanofilaria sp. Specific Genetic Assays Based on ITS2 rDNA
Amplification and Sequencing of rDNA Segment
To amplify the rDNA segment (including partial 5.8S, complete ITS2, and partial 28S) of
Stephanofilaria sp., rDNA sequences for
O. volvulus (accession no. AF228576),
M. ozzardi (accession no. MN432519),
D. repens (accession no. KP760376) and
P. bovicola (accession no. MG983750) were aligned using Geneious (version 11.1.4). Primers were designed within regions with consensus sequences targeting the desired rDNA products. The rDNA segment was amplified using a forward primer (St_5.8S_29F) located at the 3′ end of 5.8S rDNA and a reverse primer (28S_400R) located at the 5′ end of the 28S rDNA (
Table 1). Amplification and sequencing of rDNA were performed using the protocol as described above for
cox1.
All forward and reverse sequences for rDNA were aligned using Geneious (v11.1.4). Endreads and other obvious errors were corrected following inspection of the DNA sequence electropherograms. The trimmed and edited DNA sequences were used to detect similarities with other available sequences in GenBank using BLASTn (
https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 26 October 2020)).
2.8. Development and Optimisation of a Stephanofilaria sp. Specific Conventional PCR
A Stephanofilaria sp. specific primer set consisting of S_ITS2_F2 and S_ITS2_R2 was designed to amplify a 199 bp product from the ITS2 region using Primer 3 (version 4.1.0). The optimum annealing temperature for Stephanofilaria sp. specific primers was determined by setting up a gradient PCR. The PCR reaction was set up in a total volume of 20 μL containing 10 µL of 2X Phusion Hot Start II High-Fidelity PCR Master Mix (Thermo Scientific, Waltham, MA, USA, cat. no. F-565S), 10 μM of each primer, and 3 ng of DNA template. The cycling conditions included an initial denaturation at 98 °C for 30 s, followed by 30 cycles of denaturation, annealing and extension at 98 °C for 10 s, 58 ° C for 30 s and 72 °C for 30 s, respectively, and a final extension at 72 °C for 10 min. The reaction was conducted in an Eppendorf Mastercycler® pro thermal cycler (Eppendorf AG 22331 Hamburg, Germany). Gel electrophoresis was performed to ensure PCR product specificity, followed by sequencing using forward and reverse primers as described above.
2.9. Development and Optimisation of SYBR Green Quantitative (q) PCR Assay
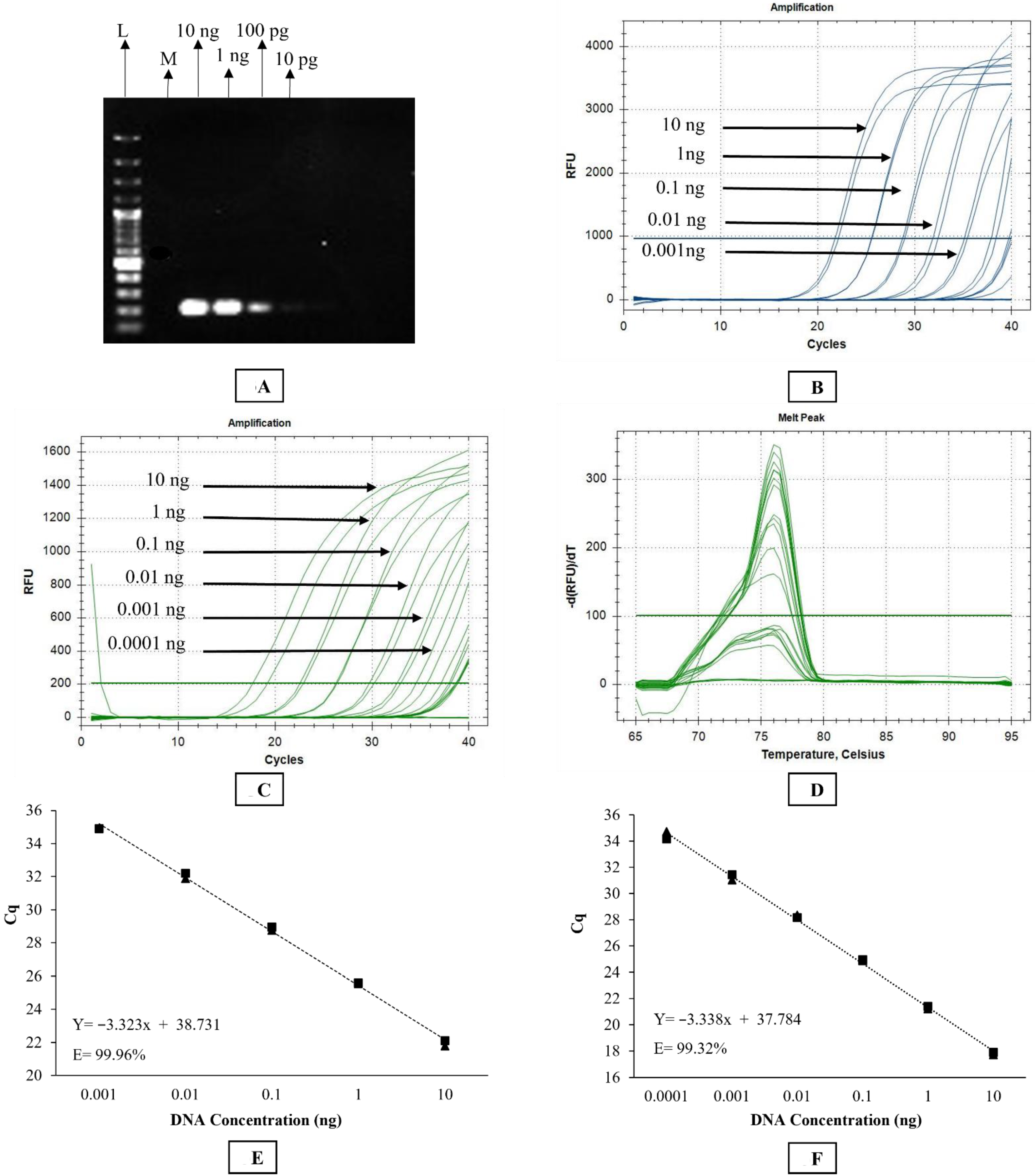
The primer set used in conventional PCR was also used for the SYBR Green-based qPCR assay. Briefly, the SYBR Green assay was conducted in a CFX96TM Real-time Detection System (Bio-Rad C1000 Touch Thermal Cycler, CALIF, Hercules, CA, USA). The reaction was set up in a total volume of 20 μL containing 10 μL of PowerUPTM SYBR Green Master Mix (Catalogue no. A25742, Thermofisher Scientific Pty, Waltham, MA, USA), 10 μM of each primer, and 1.5 ng of DNA template. The cycling conditions involved initial UDG activation at 50 °C for 2 min, Dual-lock TM DNA polymerase activation at 95 °C for 2 min, followed by 40 cycles of denaturation, annealing, and extension at 95 °C for 15 s, 58 °C for 15 s, and 72 °C for 60 s, respectively. Quantitation cycle (Cq) scores, corresponding to the cycle number at which the amplification curve intersects the threshold at 200 relative fluorescence units (RFU), were recorded for each sample. The instrument was set to perform the default melt curve analysis as described by the manufacturer. Every reaction was performed in duplicate with a dual negative control (no template control) in each qPCR run.
2.10. Development and Optimisation of TaqMan qPCR Assay
Stephanofilaria sp. specific primers (forward AP326KJ_F and reverse AP326KJ_R) and a fluorescence-labelled (FAM-labelled) probe (AP326KJ_Probe) were designed from selected ITS2 sequences using the Thermofisher Custom TaqMan™ Assay designing tool (
Table 1). The assay was ordered through Applied Biosystems (Thermofisher Scientific Pty, Waltham, MA, USA). This reaction was also conducted in a CFX96
TM Real-Time Detection System (Bio-Rad C1000 Touch Thermal Cycler, Hercules, CA, USA). The reaction was set up in a total volume of 20 µL containing 10 µL of TaqMan
TM Fast advance Master Mix (Catalogue no. 4444556, Thermofisher Scientific Pty, Waltham, MA, USA), 1 µL of 20X TaqMan assay mixture, and 1.5 ng of DNA template. The cycling conditions involved initial UNG activation at 50 °C for 2 min, AmpliTaq
TM Fast DNA polymerase activation at 95 °C for 2 min, followed by 40 cycles of denaturation, annealing/extension at 95 °C for 3 s and 60 °C for 30 s, respectively. Quantitation cycle (Cq) scores, corresponding to the cycle number at which the amplification curve intersects the threshold line at 1000 relative fluorescence units (RFU), were recorded for each sample. Every reaction was performed in duplicate with a dual negative control (no template control) in each TaqMan qPCR run.
2.11. Specificity and Sensitivity Testing
The ability of the three molecular assays to specifically amplify Stephanofilaria sp. DNA was tested against O. gibsoni and D. immitis DNA, as both species are closely related to Stephanofilaria. Onchocerca gibsoni and D. immitis DNA was acquired from the Nematode Functional Genomics Laboratory (La Trobe University, Bundoora, Victoria, Australia) and College of Public Health, Medical and Veterinary Sciences (James Cook University, Townsville, Australia), respectively. The specificity of both the conventional and qPCR assays was tested by using 2.5 ng of each DNA template.
Assay specificity was also tested against the BF vector and bovine host DNA. Laboratory reared BF (negative for Stephanofilaria sp.) kept under control conditions, were obtained from the EcoScience Precinct (Dutton Park 4102, Queensland, Australia). Skin biopsies were taken from cattle with no clinical Stephanofilaria lesions kept at the University of Queensland Pinjarra Hills research farm (UQ Animal Ethics Approval No. QAAFI469/l 8). DNA was extracted from individual BF and a cow skin biopsy using the QIAGEN DNeasy Blood and Tissue extraction kit (QIAGEN). Specificity against bovine host and fly vector DNA was tested using 2.5 ng of each DNA template in conventional and qPCRs as described above.
For sensitivity testing, DNA was extracted from 15 pooled worms using the protocol detailed above (nematode DNA extraction section). DNA was diluted 10-fold from 10 ng/µL to 1 fg/µL to determine the sensitivity of each assay.
2.12. Validation of Assays to Detect Microfilariae
To test the validity of the molecular assays for detecting DNA from microfilariae of Stephanofilaria sp., microfilariae were isolated from sediments following the saline recovery technique. DNA was extracted from single, two, and three microfilariae separately using the QIAGEN DNeasy Blood and Tissue Extraction Kit and used as a template in both conventional and qPCRs.
2.13. Detection of Stephanofilaria sp. DNA with BF Background
To ensure that the BF DNA did not interfere with the Stephanofilaria sp. DNA detection, mixtures of DNA were tested with qPCR. BF DNA was diluted 2-fold from 2400 ng/µL to 75 ng/µL, and each dilution was mixed with 1 pg/µL of Stephanofilaria sp. DNA and tested in qPCR assays. In addition, 1 pg/µL of Stephanofilaria sp. DNA and 2400 ng/ µL of BF DNA were run independently as positive and negative controls, respectively.
2.14. Preliminary Validation of Stephanofilaria sp. Specific Assays Using Bovine Skin Lesion Biopsies
The ability of all three molecular assays to detect
Stephanofilaria sp. DNA in cattle skin was validated by testing cattle skin lesion biopsies preserved in ethanol in
Section 2.1. DNA was extracted from the skin lesion biopsies using a QIAGEN DNeasy Blood and Tissue Extraction Kit and 100 ng of DNA was used as a template in both conventional and qPCR assays as described above. The findings of molecular assays were compared to the direct detection of
Stephanofilaria sp. in bovine skin lesions.
4. Discussion
In Australia, BF-associated lesions have been speculatively associated with
Stephanofilaria sp. nematode infection. However, the exact aetiology of these lesions is still unclear. In the USA, hypersensitivity induced by horn-fly feeding and the involvement of bacteria, in particular
Staphylococcus aureus, have been suggested as contributing factors in the development of horn-fly associated lesions [
28,
29]. However, due to the lower sensitivity of available techniques for detecting
Stephanofilaria sp. nematodes, investigating the pathogenesis and epidemiology of these lesions has been difficult. Therefore, to clarify the role of
Stephanofilaria sp. in the development and epidemiology of BF lesions, a more sensitive detection technique was required. This paper reports the first conventional and qPCR assays to detect different life stages of
Stephanofilaria in vector fly and definitive hosts, as well as preliminary morphological and phylogenetic analyses for this novel nematode species.
In this study, the nematodes were isolated from lesions near the medial canthus of eyes from cattle and from the same area of Queensland from which Johnson [
6] first identified
Stephanofilaria sp. in Australia. The morphology of the nematodes, geographical location, and the clinical appearance of lesions from which the nematodes were collected, as well as their detection in buffalo flies, indicates that the nematodes were similar to those described previously [
6]. The two most closely related genera of filarial nematodes described from cattle with similar cutaneous locations are
Onchocerca spp. and
Parafilaria spp. In Australia,
Onchocerca spp. are transmitted by
Culicoides marksi, Forcipomyia (Lasiohelea) townsvillensis, and
Austrosimulium pestilens [
30] and cause cutaneous nodules in cattle. The adult
Onchocerca species are much larger (up to 50 cm) than the nematodes examined in this study [
8].
Parafilaria bovicola are mainly transmitted by
Musca spp., and the adults are of similar in size to
Stephanofilaria, but they have not to date been reported from Australian cattle [
8]. In addition, gravid female nematodes isolated from the skin lesions in this study carried microfilariae enclosed in spherical vitelline membranes within the uterus, and similar membrane-enclosed microfilariae were isolated from host tissues. This is consistent with previous morphological studies of microfilariae from other species within the genus
Stephanofilaria [
6,
7,
31,
32]. In comparison,
Onchocerca spp. and
Parafilaria spp. have unsheathed microfilariae [
24,
30].
Scanning electron micrographs also suggest that the nematodes in this study are the same species as those reported in the past [
6]. The only differences in the morphological features described in this study were in the number of peri-buccal spines observed and the presence of cuticular spines. Johnson [
6], using light microscopy, observed 15–16 peri-buccal spines, whereas 18 were counted using SEM in our study. In addition, Johnson [
6] reported no cuticular spines, while cuticles with spines directed both anteriorly and posteriorly were observed in this study. It is difficult to count the exact number of peri-buccal spines using light microscopy, and Johnson [
6] noted that counting of peri-buccal spines was imprecise, as the spines were difficult to distinguish from the supporting collar. This difference is relatively minor, and the higher number of peri-buccal spines noted here is likely the result of the clearer definition possible using SEM. In common with previous observations, no cephalic spines were observed which suggests that this characteristic may differentiate the Australian
Stephanofilaria sp. [
6] from
S. stilesi vectored by
H. irritans which was reported to have 4–5 cephalic spines [
7]. Further determination of the species status of the Australian
Stephanofilaria and
S. stilesi will require a more detailed morphological and molecular comparison of the two morphotypes.
Mitochondrial DNA is genetically conserved within species in the same genus, and
cox1 sequences have thus been used previously in many studies to reveal phylogenetic relationships among species [
24,
33,
34,
35,
36,
37]. In addition, rDNA (ITS1 and ITS2) has been previously used for delineation and identification of filarial nematodes [
17,
38]. Thus, amplification and sequencing of the
cox1 gene were used for the determination of the phylogenetic relationship with other representing species closely related to
Stephanofilaria sp. The ITS2 region was chosen as a suitable target for use in specific diagnostic assays for
Stephanofilaria sp. Internal transcribed spacer (ITS)-based PCR assays have already been reported for specific detection of different nematodes from order Strongylida, Ascaridida, and Spirurida [
16,
18,
19,
20,
21,
22,
23,
24].
From the BLASTn sequence comparison, the
cox1 and partial rDNA sequences of
Stephanofilaria sp. showed maximum similarities to
Dirofilaria,
Onchocerca,
Thelazia, and
Loa spp. At first glance, this was not surprising, as the genus
Stephanofilaria is classified within the superfamily Filarioidea [
39]. Among the most similar genera from this group,
Dirofilaria and
Onchocerca belong to the family Onchocercidae, but
Thelazia belongs to the family Thelaziidae. However, the genus
Stephanofilaria is currently placed in the family Filariidae. For the Filariidae,
cox1 and rDNA sequences hitherto have been available in GenBank for species of
Parafilaria,
Brugia, and
Loa. Of these, members of the genus
Loa showed the highest similarity (83.68%) with
Stephanofilaria sp. Overall, no other member of the family Filariidae was among the top 20 BLASTn hits. Phylogenetic analysis in this study indicated that
Stephanofilaria sp. shares a closer common ancestor with
T. callipaeda than with any member of the family Filariidae. This close association is of particular interest given the similar biology and close proximity of infestations of
T. gulosa and
T. skrjabini in Australian cattle [
40]. The
Stephanofilaria sp. nematodes used in this study were collected from lesions on the medial canthus of the eye, whereas
Thelazia spp., previously isolated from Australian cattle, were found in the conjunctival sac, under the third eyelid and within the lachrymal ducts and nasolachrymal canal [
40]. Interestingly,
Stephanofilaria has been moved between families in the order Spirurida on several occasions based on its unique morphological and biological characteristics and the predilection site of the nematode [
41]. Boomker et al. [
2] isolated
Stephanofilaria nematodes from the skin of a hippopotamus which he named
Stephanofilaria thelazioides n. sp because of morphological similarities to the genus
Thelazia. The results of this study may suggest that
Stephanofilaria should be placed in the family Thelaziidae but more conserved
Stephanofilaria genes, and more sequences from other
Stephanofilaria species and their close relatives are required to support this claim.
The rDNA sequence from our study contained the complete ITS2 and partial 28S segment. The ITS2 sequence of
Stephanofilaria sp. had no similarity with any sequence available in GenBank. Due to the greater divergence in the ITS2, it was used to design species-specific conventional and qPCR assays for the identification and delineation of both adult and larval stages of
Stephanofilaria sp. Both conventional and qPCR assays specifically amplified
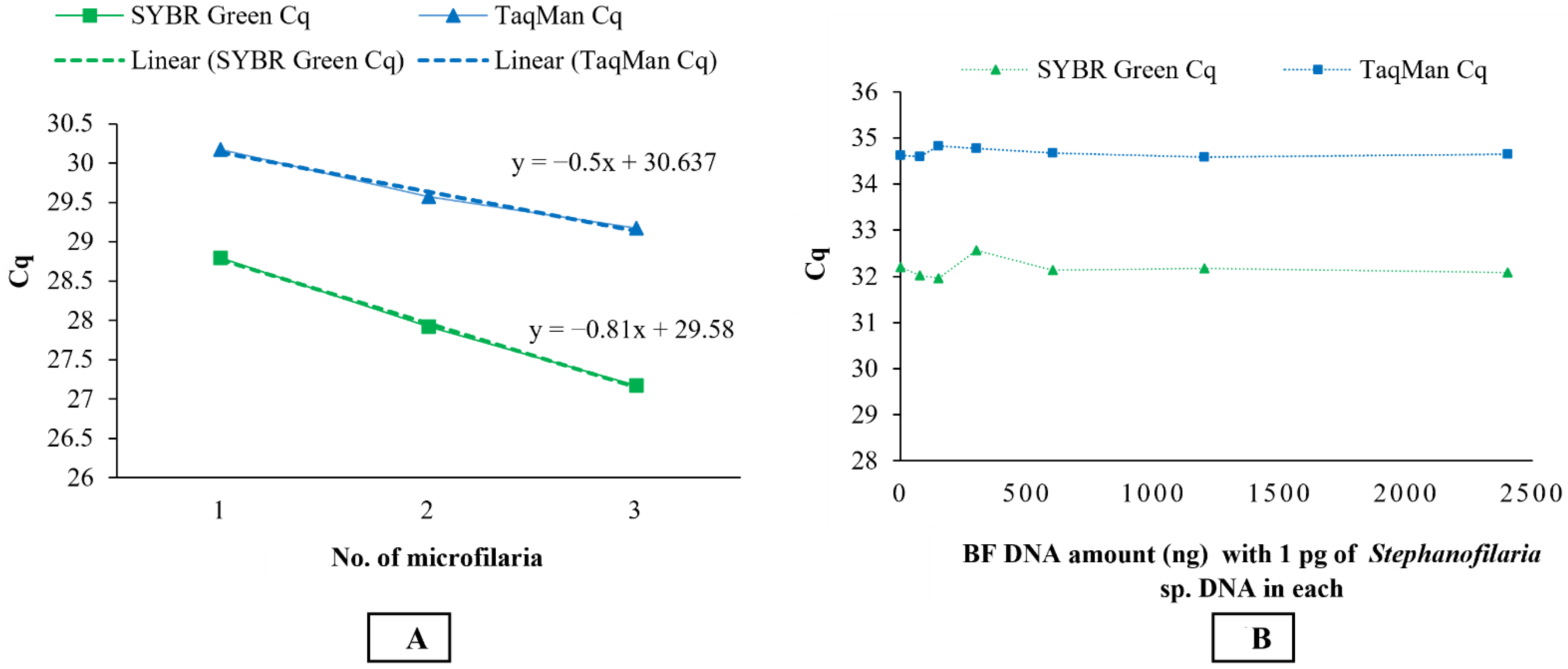
Stephanofilaria sp. DNA with no amplification for other filarial nematode DNA tested in these studies. Comparison of the Cq values from microfilariae DNA to the standard curve for SYBR Green and TaqMan qPCR indicated that individual microfilaria yields a low amount of DNA (150–200 pg) and the lower number of cycles we used for the conventional PCR (30 cycles), compared to qPCR assays (40 cycles), significantly affected the assay sensitivity. Quantitative PCR assays are highly sensitive and relatively advanced tools for high-throughput detection of target DNA and are rapidly replacing conventional PCR and microscopic methods as detection methods for parasites. Both the SYBR Green and TaqMan assays developed in this study were able to detect DNA from individual microfilaria with a reliable Cq (<31). Overall, the SYBR Green assay appeared to be 10 times more sensitive than the TaqMan assay, and this has also been observed in the detection of
Babesia spp. [
42]. However, melting temperature analysis is needed to ensure specificity of the SYBR Green assay and this could be difficult under some circumstances. This suggests that the TaqMan assay may be more suitable for general use to ensure cleaner results and more robust diagnostic specificity.
The significantly higher sensitivity of PCR-based assays for nematode detection compared to traditional histological methods has been reported in previous studies with other nematode species [
16,
43,
44]. Similarly, our findings showed that molecular assays were more sensitive than saline recovery detection. Although the qPCR technique detected as little as 100 fg of
Stephanofilaria sp. DNA, the sensitivity of detection of
Stephanofilaria sp. in the skin lesions, will also be strongly dependent on the sampling methods used. The higher sensitivity of the qPCR assays may also allow detection from non-invasive samples (e.g., from lesion swabs), as there is evidence of the presence of skin infecting nematodes and microfilariae at the surface of active lesions [
24]. Quantitative PCR assays detected nematode DNA in four skin samples which were negative for the presence of nematode or microfilariae using the saline recovery technique. However, it is yet to be confirmed whether molecular detection relies on the presence of whole or partial nematodes or microfilariae in the sample collected, or whether the PCR may be able to detect the presence of
Stephanofilaria sp. from secretory or excretory compounds or cellular material abraded during nematode migration.
This study also showed that there was no effect on the sensitivity of qPCR assays in detecting 1 pg of
Stephanofilaria sp. DNA with high background concentrations (2400 ng/ µL) of buffalo fly DNA. An amount of 1 pg is approximately 0.5–0.66% of DNA found in single microfilaria, suggesting that both SYBR Green and TaqMan assays could be used to detect first-, second-, and third-stage
Stephanofilaria sp. larvae in BF. Quantitative PCR-based detection techniques have also been used to detect
Brugia malayi and
Wuchereria bancrofti in mosquitoes [
45,
46]. The PCR-based assays developed here would overcome the need for time-consuming dissection and examination of BF to determine the presence of infection. Furthermore, these assays may also have application for use in epidemiological studies with other species of
Stephanofilaria in other vector and host associations, especially closely related
S. stilesi, but this requires confirmation.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}