
Giardia duodenalis in Wildlife: Exploring Genotype Diversity in Italy and across Europe

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Genotyping of the Italian Samples at the SSU-rRNA Locus
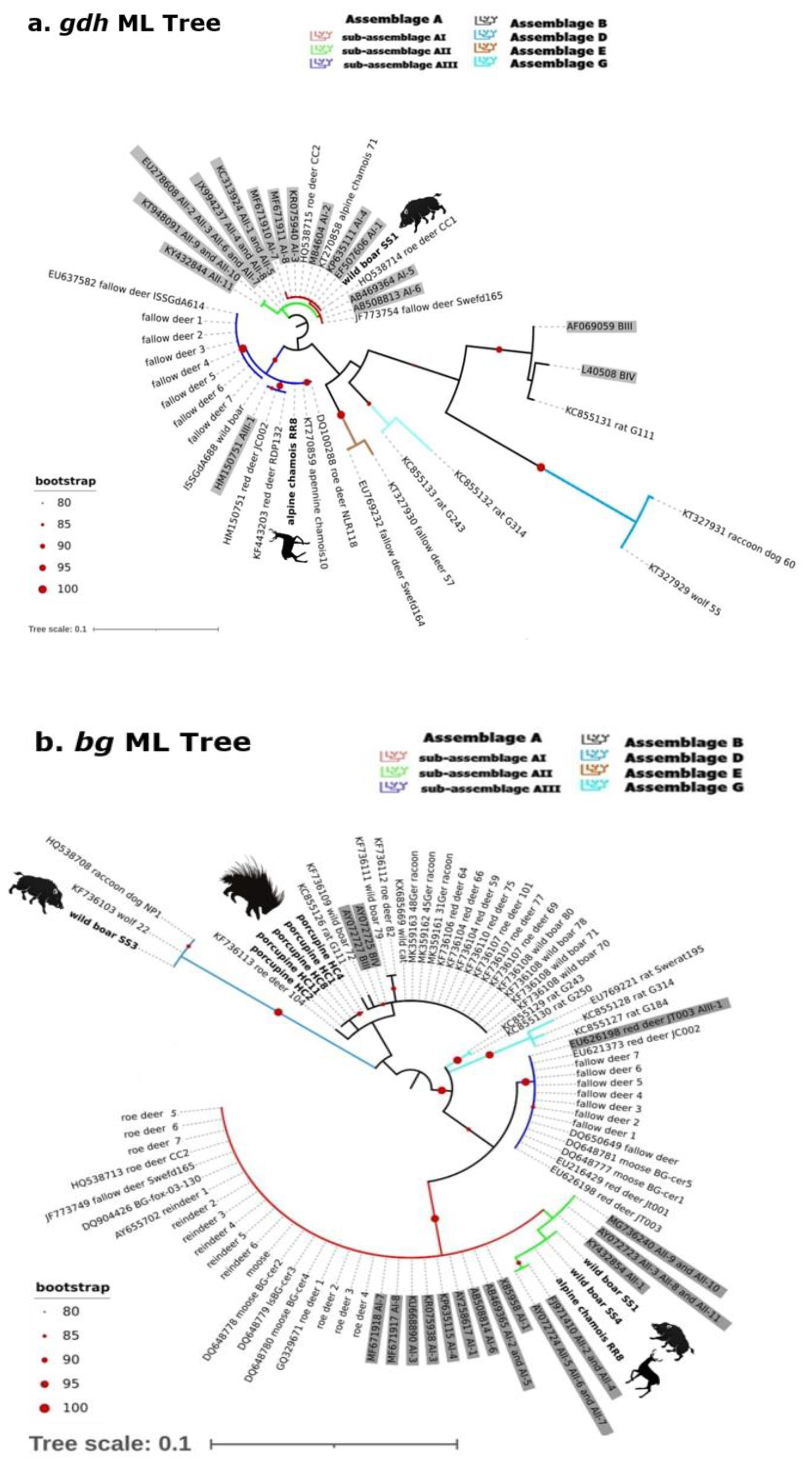
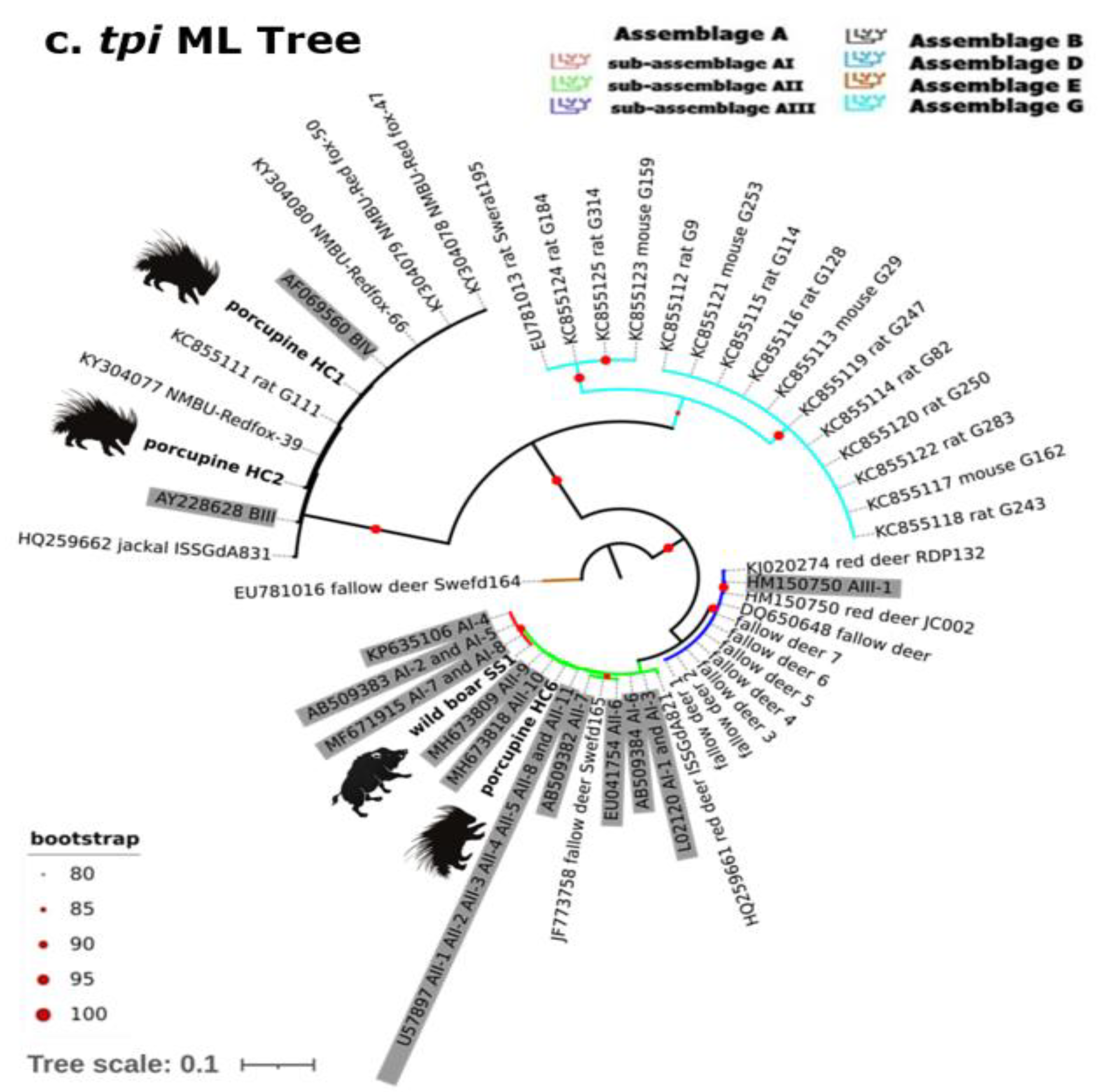
2.2. Gdh, Bg and Tpi Phylogenetic Analysis
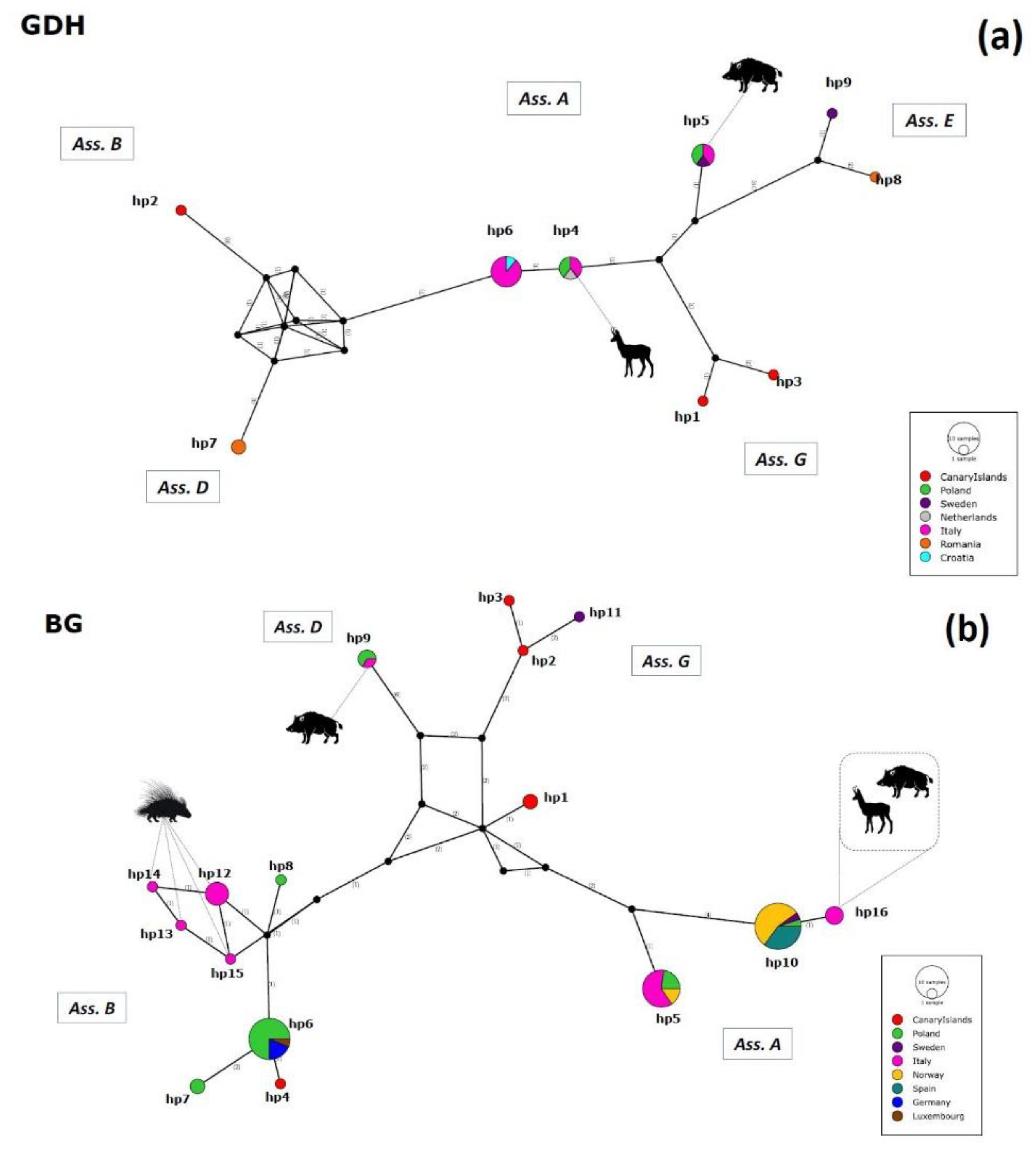
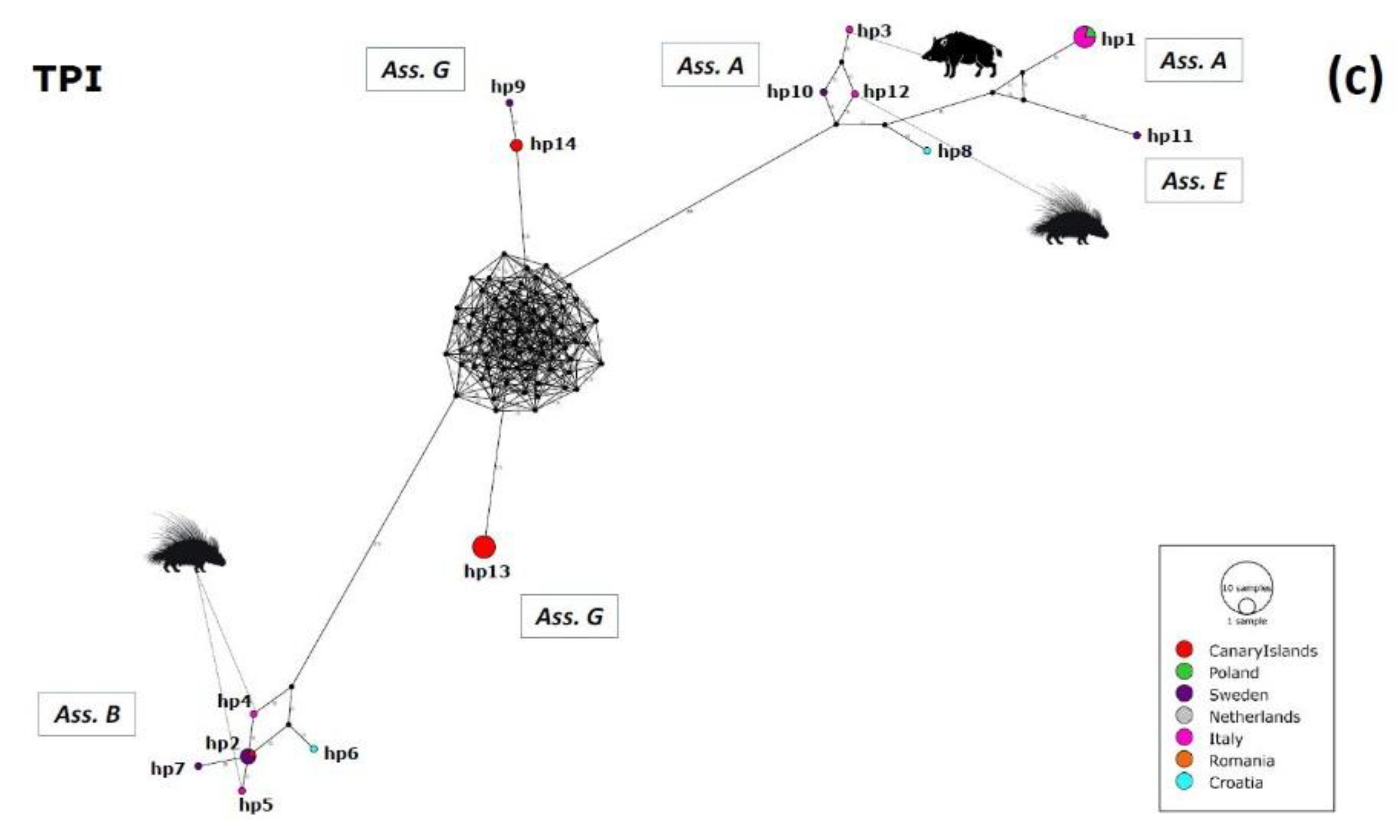
2.3. Haplotype Variability
3. Discussion
4. Materials and Methods
4.1. Sampling
4.2. Assemblage and Sub-Assemblage Identification
4.3. Haplotype Analysis and Networks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Hudson, P.J.; Rizzoli, A.P.; Grenfell, B.T.; Heesterbeek, H.; Dobson, A.P. The Ecology of Wildlife Diseases; Oxford University Press: Oxford, UK, 2002; p. 197. [Google Scholar]
- Gómez, A.; Nichols, E. Neglected wild life: Parasitic biodiversity as a conservation target. Int. J. Parasitol. Parasites Wildl. 2013, 2, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, A.P.; Linklater, W. Everything in Moderation: Principles of Parasite Control for Wildlife Conservation. BioScience 2014, 64, 932–939. [Google Scholar] [CrossRef] [Green Version]
- Hudson, P.J.; Dobson, A.P.; Lafferty, K.D. Is a healthy ecosystem one that is rich in parasites? Trends Ecol. Evol. 2006, 21, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, K.D.; Allesina, S.; Arim, M.; Briggs, C.J.; De Leo, G.; Dobson, A.P.; Dunne, J.A.; Johnson, P.T.; Kuris, A.M.; Marcogliese, D.J.; et al. Parasites in food webs: The ultimate missing links. Ecol. Lett. 2008, 11, 533–546. [Google Scholar] [CrossRef] [PubMed]
- De Castro, F.; Bolker, B. Mechanisms of disease-induced extinction. Ecol. Lett. 2005, 8, 117–126. [Google Scholar] [CrossRef]
- Heard, M.J.; Smith, K.F.; Ripp, K.J.; Berger, M.; Chen, J.; Dittmeier, J.; Goter, M.; McGarvey, S.T.; Ryan, E. The threat of disease increases as species move toward extinction. Conserv. Biol. 2013, 27, 1378–1388. [Google Scholar] [CrossRef] [Green Version]
- Adam, R.D. Giardia duodenalis: Biology and Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00024-19. [Google Scholar] [CrossRef]
- Feng, Y.; Xiao, L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 2011, 24, 110–140. [Google Scholar] [CrossRef] [Green Version]
- Cacciò, S.M.; Lalle, M.; Svärd, S.G. Host specificity in the Giardia duodenalis species complex. Infect. Genet. Evol. 2018, 66, 335–345. [Google Scholar] [CrossRef]
- Seabolt, M.H.; Konstantinidis, K.T.; Roellig, D.M. Hidden Diversity within Common Protozoan Parasites as Revealed by a Novel Genotyping Scheme. Appl. Environ. Microbiol. 2021, 87, e02275-20. [Google Scholar] [CrossRef]
- Hulme, P.E. Invasive species challenge the global response to emerging diseases. Trends Parasitol. 2014, 3, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.J.; Forberg, T.; Hermansen, L.; Hamnes, I.S.; Gjerde, B. Giardia duodenalis cysts isolated from wild moose and reindeer in Norway: Genetic characterization by PCR-rflp and sequence analysis at two genes. J. Wildl. Dis. 2007, 43, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Beck, R.; Sprong, H.; Lucinger, S.; Pozio, E.; Caccio, S.M. A large survey of Croatian wild mammals for Giardia duodenalis reveals a low prevalence and limited zoonotic potential. Vector Borne Zoonotic Dis. 2011, 11, 1049–1055. [Google Scholar] [CrossRef]
- Stojecki, K.; Sroka, J.; Cacciò, S.M.; Cencek, T.; Dutkiewicz, J.; Kusyk, P. Prevalence and molecular typing of Giardia duodenalis in wildlife from eastern Poland. Folia Parasitol. 2015, 62, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debenham, J.J.; Landuyt, H.; Troell, K.; Tysnes, K.; Robertson, L.J. Occurrence of Giardia in Swedish Red Foxes (Vulpes vulpes). J. Wildl. Dis. 2017, 53, 649–652. [Google Scholar] [CrossRef]
- Helmy, Y.A.; Spierling, N.G.; Schmidt, S.; Rosenfeld, U.M.; Reil, D.; Imholt, C.; Jacob, J.; Ulrich, R.G.; Aebischer, T.; Klotz, C. Occurrence and distribution of Giardia species in wild rodents in Germany. Parasit. Vectors 2018, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Lalle, M.; Frangipane di Regalbono, A.; Poppi, L.; Nobili, G.; Tonanzi, D.; Pozio, E.; Cacciò, S.M. A novel Giardia duodenalis assemblage A subtype in fallow deer. J. Parasitol. 2007, 93, 426–428. [Google Scholar] [CrossRef]
- Cacciò, S.M.; Beck, R.; Lalle, M.; Marinculic, A.; Pozio, E. Multilocus genotyping of Giardia duodenalis reveals striking differences between assemblages A and B. Int. J. Parasitol. 2008, 38, 1523–1531. [Google Scholar] [CrossRef]
- De Liberato, C.; Berrilli, F.; Marangi, M.; Santoro, M.; Trogu, T.; Putignani, L.; Lanfranchi, P.; Ferretti, F.; D’Amelio, S.; Giangaspero, A. Giardia duodenalis in Alpine (Rupicapra rupicapra rupicapra) and Apennine (Rupicapra pyrenaica ornata) chamois. Parasit. Vectors 2015, 8, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, F.; Maestrini, M.; Berrilli, F.; Guadano Procesi, I.; Felicioli, A.; Perrucci, S. First report of Giardia duodenalis infection in the crested porcupine (Hystrix cristata L., 1758). Int. J. Parasitol. Parasites Wildl. 2020, 16, 108–113. [Google Scholar] [CrossRef]
- Fernández-Álvarez, Á.; Martín-Alonso, A.; Abreu-Acosta, N.; Feliu, C.; Hugot, J.P.; Valladares, B.; Foronda, P. Identification of a novel assemblage G subgenotype and a zoonotic assemblage B in rodent isolates of Giardia duodenalis in the Canary Islands, Spain. Parasitology 2014, 141, 206–215. [Google Scholar] [CrossRef]
- Solarczyk, P.; Majewska, A.C.; Moskwa, B.; Cabaj, W.; Dabert, M.; Nowosad, P. Multilocus genotyping of Giardia duodenalis isolates from red deer (Cervus elaphus) and roe deer (Capreolus capreolus) from Poland. Folia Parasitol. 2012, 59, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriana, G.; Zsuzsa, K.; Mirabela Oana, D.; Mircea, G.C.; Viorica, M. Giardia duodenalis genotypes in domestic and wild animals from Romania identified by PCR-RFLP targeting the gdh gene. Vet. Parasitol. 2016, 217, 71–75. [Google Scholar] [CrossRef]
- Lebbad, M.; Mattsson, J.G.; Christensson, B.; Ljungström, B.; Backhans, A.; Andersson, J.O.; Svärd, S.G. From mouse to moose: Multilocus genotyping of Giardia isolates from various animal species. Vet. Parasitol. 2010, 168, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Van der Giessen, J.W.B.; De Vries, A.; Roos, M.; Wielinga, P.; Kortbeek, L.M.; Mank, T.G. Genotyping of Giardia in Dutch patients and animals: A phylogenetic analysis of human and animal isolates. Int. J. Parasitol. 2006, 36, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Hamnes, I.S.; Gjerde, B.K.; Forberg, T.; Robertson, L.J. Occurrence of Giardia and Cryptosporidium in Norwegian red foxes (Vulpes vulpes). Vet. Parasitol. 2007, 143, 347–353. [Google Scholar] [CrossRef]
- García-Presedo, I.; Pedraza-Díaz, S.; González-Warleta, M.; Mezo, M.; Gómez-Bautista, M.; Ortega-Mora, L.M.; Castro-Hermida, J.A. The first report of Cryptosporidium bovis, C. ryanae and Giardia duodenalis sub-assemblage A-II in roe deer (Capreolus capreolus) in Spain. Vet. Parasitol. 2013, 197, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Solarczyk, P.; Dabert, M.; Frantz, A.C.; Osten-Sacken, N.; Trzebny, A.; Wojtkowiak-Giera, A.; Heddergott, M. Zoonotic Giardia duodenalis sub-assemblage BIV in wild raccoons (Procyon lotor) from Germany and Luxembourg. Zoonoses Public Health 2021, 68, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Solarczyk, P.; Osten-Sacken, N.; Frantz, A.C.; Schneider, S.; Pir, J.B.; Heddergott, M. First molecular detection of Giardia duodenalis assemblage B in a free-living European wildcat (Felis s. silvestris) from Luxembourg. Acta Protozool. 2019, 58, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Trout, J.M.; Santín, M.; Greiner, E.; Fayer, R. Prevalence of Giardia duodenalis genotypes in pre-weaned dairy calves. Vet. Parasitol. 2004, 124, 179–186. [Google Scholar] [CrossRef]
- Lebbad, M.; Petersson, I.; Karlsson, L.; Botero-Kleiven, S.; Andersson, J.O.; Svenungsson, B.; Svärd, S.G. Multilocus genotyping of human Giardia isolates suggests limited zoonotic transmission and association between assemblage B and flatulence in children. PLoS Negl. Trop. Dis. 2011, 5, e1262. [Google Scholar] [CrossRef] [Green Version]
- Di Francesco, C.E.; Smoglica, C.; Paoletti, B.; Angelucci, S.; Innocenti, M.; Antonucci, A.; Di Domenico, G.; Marsilio, F. Detection of selected pathogens in Apennine wolf (Canis lupus italicus) by a non-invasive GPS-based telemetry sampling of two packs from Majella National Park, Italy. Eur. J. Wildl. Res. 2019, 65, 84. [Google Scholar] [CrossRef]
- Mizuno, T.; Matey, E.J.; Bi, X.; Songok, E.M.; Ichimura, H.; Tokoro, M. Extremely diversified haplotypes observed among assemblage B population of Giardia intestinalis in Kenya. Parasitol. Int. 2020, 75, 102038. [Google Scholar] [CrossRef]
- Garcia-R, J.C.; French, N.; Pita, A.; Velathanthiri, N.; Shrestha, R.; Hayman, D. Local and global genetic diversity of protozoan parasites: Spatial distribution of Cryptosporidium and Giardia genotypes. PLoS Negl. Trop. Dis. 2017, 11, e0005736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spotin, A.; Karamat, M.; Mahami-Oskouei, M.; Shahbazi, A.; Ahmadpour, E.; Galeh, T.M.; Fallahi, S. Genetic variability and transcontinental sharing of Giardia duodenalis infrapopulations determined by glutamate dehydrogenase gene. Acta Trop. 2018, 177, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Capewell, P.; Krumrie, S.; Katzer, F.; Alexander, C.L.; Weir, W. Molecular epidemiology of Giardia infections in the Genomic Era. Trends Parasitol. 2021, 37, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Read, C.; Walters, J.; Robertson, I.D.; Thompson, R.C. Correlation between genotype of Giardia duodenalis and diarrhoea. Int. J. Parasitol. 2002, 32, 229–231. [Google Scholar] [CrossRef]
- Read, C.M.; Monis, P.T.; Thompson, R.C. Discrimination of all genotypes of Giardia duodenalis at the glutamate dehydrogenase locus using PCR-RFLP. Infect. Genet. Evol. 2004, 4, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Cacciò, S.M.; De Giacomo, M.; Pozio, E. Sequence analysis of the beta-giardin gene and development of a polymerase chain reaction-restriction fragment length polymorphism assay to genotype Giardia duodenalis cysts from human faecal samples. Int. J. Parasitol. 2002, 32, 1023–1030. [Google Scholar] [CrossRef]
- Sulaiman, I.M.; Fayer, R.; Bern, C.; Gilman, R.H.; Trout, J.M.; Schantz, P.M.; Das, P.; Lal, A.A.; Xiao, L. Triosephosphate isomerase gene characterization and potential zoonotic transmission of Giardia duodenalis. Emerg. Infect. Dis. 2003, 9, 1444–1452. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Ryan, U.; Xiao, L.; Feng, Y. Zoonotic giardiasis: An update. Parasitol. Res. 2021, 120, 4199–4218. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M.; Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Species | Origin | Specimen Code | Assemblage/ Sub-Assemblage (n) | GenBank Accession Number | Ref. |
|---|---|---|---|---|---|
| SSU_rRNA | |||||
| Rupicapra r. rupicapra | “ | RR6; RR7; RR8; RR9; RR11; RR12 | A(3); E(2); A+E(1) | OL840340-42 | present study |
| Canis lupus | “ | CL1 | D | OL840343 | present study |
| Sus scrofa | “ | SS1-SS4 | A(4) | OL840344 | present study |
| Hystrix cristata | “ | HC1-HC12 | B(12) | OL840345 | present study |
| Dataset I gdh | |||||
| Rupicapra r. rupicapra | “ | RR8 | AIII | OL828750 | present study |
| Sus scrofa | “ | SS1 | AI | OL828751 | present study |
| Rattus rattus | Spain: Canary Islands | G111; G314; G243 | B(1); G(2) | KC855131-33 | Fernández-Álvarez et al., 2014 [22] |
| Cervus elpahus | Poland | JC002 | A | HM150751 | Solarczyk et al., 2012 [23] |
| Cervus elpahus | “ | RDP132 | A | KF443203 | unpublished |
| Capreolus capreolus | “ | CC1; CC2 | A(2) | HQ538714-15 | Solarczyk et al., 2012 [23] |
| Sus scrofa | Croatia | ISSGdA688 | A | sequence not deposited 1 | Beck et al., 2011 [14] |
| Canis lupus | Romania | wolf55 | D | KT327929 | Adriana et al. 2016 [24] |
| Dama dama | “ | deer57 | E | KT327930 | “ |
| Nyctereutes procyonoides | “ | racoondog60 | D | KT327931 | “ |
| Dama dama | Sweden | Swefd164; Sewfd165 | E(1); A(1) | EU769232; JF773754 | Lebbad et al., 2010 [25] |
| Capreolus capreolus | The Netherlands | NLR118 | A | DQ100288 | van der Giessen et al., 2006 [26] |
| Dama dama | Italy | ISSGdA614;(fallow deer 1–7) | AIII(8) | EU637582 | Cacciò et al., 2008 [19] |
| Rupicapra r. rupicapra | “ | 71 | AI | KT270858 | De Liberato et al., 2015 [20] |
| Rupicapra p. ornata | “ | 10 | AIII | KT270859 | “ |
| Dataset II bg | |||||
| Sus scrofa | Italy | SS1; SS4; SS3 | AII(2) D(1) | OL944445-46 | present study |
| Hystrix cristata | “ | HC1; HC2; HC4; HC8; HC11 | B(5) | OL944442-44 | present study |
| Rupicapra r. rupicapra | “ | RR8 | AII | OL944447 | present study |
| Rattus rattus | Spain: Canary Island | G111; G184; G314; G243; G250 | B(1); G(4) | KC855126-30 | Fernández-Álvarez et al., 2014 [22] |
| Cervus elaphus | Poland | JC002 | A | EU621373; | Solarczyk et al., 2012 [23] |
| Capreolus capreolus | “ | CC2 | A | HQ538713 | Solarczyk et al., 2012 [23] |
| Cervus elaphus | Poland | JT001, JT003 | A(2) | EU216429; EU626198 | unpublished |
| Nyctereutes procyonoides | “ | NP1 | D | HQ538708 | unpublished |
| Sus scrofa | “ | 70; 71; 78; 80; 72; 79 | B(6) | KF736108-09; KF736111 | Stojecki et al., 2015 [15] |
| Capreolus capreolus | “ | 69; 77; 101; 82; 104 | B(5) | KF736107; KF736112-13 | “ |
| Canis lupus | “ | 22 | D | KF736103 | “ |
| Cervus elaphus | “ | 59, 66; 64; 75 | B(4) | KF736104; KF736106; KF736110 | “ |
| Rattus norvergicus | Sweden | Swerat195 | G | EU769221 | Lebbad et al., 2010 [25] |
| Dama dama | “ | Swefd165 | A | JF773749 | “ |
| Vulpes vulpes | Norway | BG-fox-03-130 | A | DQ904426 | Hamnes et al., 2007 [27] |
| Rangifer tarandus | “ | (reindeer 1–6) | A(6) | sequence not deposited 2 | Robertson et al., 2007 [13] |
| Alces alces | “ | (moose) | A | sequence not deposited 2 | “ |
| Alces alces | “ | BG-cer1; BG-cer2; BG-cer3; BG-cer4;BG-cer5 | A(5) | DQ648777-81 | “ |
| Capreolus capreolus | Spain | (roe deer 1–7) | AII(7) | sequence not deposited 3 | García-Presedo et al., 2013 [28] |
| Dama dama | Italy | (fallow deer 1–8) | AIII(8) | DQ650649 | Lalle et al., 2007 [18] |
| Procyon lotor | Germany | 31; 45; 48 | BIV(3) | MK359161-63 | Solarczyk et al., 2021 [29] |
| Felis silvestris | Luxembourg | FS1 | B | KX685669 | Solarczyk et al., 2019 [30] |
| Dataset III tpi | |||||
| Sus scrofa | Italy | SS1 | AI | OL944434 | present study |
| Hystrix cristata | “ | HC1; HC2; HC6; | BIV(2); AII(1) | OL944435-37 | present study |
| Rattus rattus | Spain: Canary Island | G111; G9; G82; G114; G128; G243; G247; G250; G283; G184; G314 | B(1); G(10) | KC855111-12; KC855114-16; KC855118-20; KC855122; KC855124-25 | Fernández-Álvarez et al., 2014 [22] |
| Mus musuculus domesticus | “ | G29; G162; G253; G159 | G(4) | KC855113; KC855117; KC855121; KC855123 | “ |
| Cervus elaphus | Poland | JC002 | A | HM150750 | Solarczyk et al., 2012 [23] |
| Cervus elaphus | “ | RDP132 | A | KJ020274 | unpublished |
| Cervus elaphus | Croatia | ISSGdA821 | A | HQ259661 | Beck et al., 2011 [14] |
| Canis aureus moreoticus | “ | ISSGdA831 | B | HQ259662 | “ |
| Rattus norvegicus | Sweden | Swerat195 | G | EU781013 | Lebbad et al., 2010 [25] |
| Dama dama | “ | Swefd164; Swefd165 | E(1); A(1) | EU781016; JF773758 | “ |
| Vulpes vulpes | “ | NMBU-Red fox-39; NMBU-Red fox-47; NMBU-Red fox-50; NMBU-Red fox-66 | B(4) | KY304077-80 | Debenham et al., 2017 [16] |
| Dama dama | Italy | (fallow deer 1–8) | AIII(8) | DQ650648 | Lalle et al., 2007 [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guadano Procesi, I.; Montalbano Di Filippo, M.; De Liberato, C.; Lombardo, A.; Brocherel, G.; Perrucci, S.; Di Cave, D.; Berrilli, F. Giardia duodenalis in Wildlife: Exploring Genotype Diversity in Italy and across Europe. Pathogens 2022, 11, 105. https://doi.org/10.3390/pathogens11010105
Guadano Procesi I, Montalbano Di Filippo M, De Liberato C, Lombardo A, Brocherel G, Perrucci S, Di Cave D, Berrilli F. Giardia duodenalis in Wildlife: Exploring Genotype Diversity in Italy and across Europe. Pathogens. 2022; 11(1):105. https://doi.org/10.3390/pathogens11010105
Chicago/Turabian StyleGuadano Procesi, Isabel, Margherita Montalbano Di Filippo, Claudio De Liberato, Andrea Lombardo, Giuseppina Brocherel, Stefania Perrucci, David Di Cave, and Federica Berrilli. 2022. "Giardia duodenalis in Wildlife: Exploring Genotype Diversity in Italy and across Europe" Pathogens 11, no. 1: 105. https://doi.org/10.3390/pathogens11010105
APA StyleGuadano Procesi, I., Montalbano Di Filippo, M., De Liberato, C., Lombardo, A., Brocherel, G., Perrucci, S., Di Cave, D., & Berrilli, F. (2022). Giardia duodenalis in Wildlife: Exploring Genotype Diversity in Italy and across Europe. Pathogens, 11(1), 105. https://doi.org/10.3390/pathogens11010105