
Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis


Abstract
:1. Introduction
2. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeyaullah, M.; AlShahrani, A.M.; Muzammil, K.; Ahmad, I.; Alam, S.; Khan, W.H.; Ahmad, R. COVID-19 and SARS-CoV-2 Variants: Current Challenges and Health Concern. Front. Genet. 2021, 12, 693916. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; O’Donnell, A.C.; Ochman, H. Recombination events are concentrated in the spike protein region of Betacoronaviruses. PLoS Genet. 2020, 16, e1009272. [Google Scholar] [CrossRef]
- Chakraborty, S. Evolutionary and structural analysis elucidates mutations on SARS-CoV2 spike protein with altered human ACE2 binding affinity. Biochem. Biophys. Res. Commun. 2021, 534, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [CrossRef]
- Mengist, H.M.; Kombe, A.J.K.; Mekonnen, D.; Abebaw, A.; Getachew, M.; Jin, T. Mutations of SARS-CoV-2 spike protein: Implications on immune evasion and vaccine-induced immunity. Semin. Immunol. 2021, 55, 101533. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Parashar, R.; Kumar, S.; Faiq, M.A.; Kumari, C.; Kulandhasamy, M.; Narayan, R.K.; Jha, R.K.; Singh, H.N.; Prasoon, P.; et al. Emerging SARS-CoV-2 variants can potentially break set epidemiological barriers in COVID-19. J. Med. Virol. 2021, 1–15. [Google Scholar] [CrossRef]
- Pyke, A.T.; Nair, N.; van den Hurk, A.F.; Burtonclay, P.; Nguyen, S.; Barcelon, J.; Kistler, C.; Schlebusch, S.; McMahon, J.; Moore, F. Replication Kinetics of B.1.351 and B.1.1.7 SARS-CoV-2 Variants of Concern Including Assessment of a B.1.1.7 Mutant Carrying a Defective ORF7a Gene. Viruses 2021, 13, 1087. [Google Scholar] [CrossRef]
- Ahmad, L. Implication of SARS-CoV-2 Immune Escape Spike Variants on Secondary and Vaccine Breakthrough Infections. Front. Immunol. 2021, 12, 742167. [Google Scholar] [CrossRef]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage–United States, 29 December 2020–12 January 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef]
- Zhang, Y.; He, X.; Zhai, J.; Ji, B.; Man, V.H.; Wang, J. In silico binding profile characterization of SARS-CoV-2 spike protein and its mutants bound to human ACE2 receptor. Brief. Bioinform. 2021, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; Yousafi, Q.; Ali, J.; Ali, S.S.; Hussain, Z.; Ali, S.; Waseem, M.; Iqbal, A.; Ahmad, S.; Khan, A.; et al. Bioinformatics analysis of the differences in the binding profile of the wild-type and mutants of the SARS-CoV-2 spike protein variants with the ACE2 receptor. Comput. Biol. Med. 2021, 138, 104936. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.T.; Pujol, F.H.; Jastrzebska, B.; Rangel, H.R. Mutations in the SARS-CoV-2 spike protein modulate the virus affinity to the human ACE2 receptor, an in silico analysis. EXCLI J. 2021, 20, 585–600. [Google Scholar] [CrossRef]
- Hodcroft, E.B. CoVariance: SARS-CoV-2 Mutations and Variants of Interest. 2021. Available online: https://covariants.org/ (accessed on 26 November 2021).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Rangel, H.R.; Ortega, J.T.; Maksoud, S.; Pujol, F.H.; Serrano, M.L. SARS-CoV-2 host tropism: An in silico analysis of the main cellular factors. Virus Res. 2020, 289, 198154. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Moss, D.S.; Thornton, J.M. Main-Chain Bond Lengths and Bond Angles in Protein Structures. J. Mol. Biol. 1993, 231, 1049–1067. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI J. 2020, 19, 400–409. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Jastrzebska, B. Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis. Biomolecules 2020, 10, 954. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiros-Reis, L.; Gomes da Silva, P.; Goncalves, J.; Brancale, A.; Bassetto, M.; Mesquita, J.R. SARS-CoV-2 Virus-Host Interaction: Currently Available Structures and Implications of Variant Emergence on Infectivity and Immune Response. Int. J. Mol. Sci. 2021, 22, 10836. [Google Scholar] [CrossRef]
- Li, T.; Han, X.; Gu, C.; Guo, H.; Zhang, H.; Wang, Y.; Hu, C.; Wang, K.; Liu, F.; Luo, F.; et al. Potent SARS-CoV-2 neutralizing antibodies with protective efficacy against newly emerged mutational variants. Nat. Commun. 2021, 12, 6304. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Chen, C.; Boorla, V.S.; Banerjee, D.; Chowdhury, R.; Cavener, V.S.; Nissly, R.H.; Gontu, A.; Boyle, N.R.; Vandegrift, K.; Nair, M.S.; et al. Computational prediction of the effect of amino acid changes on the binding affinity between SARS-CoV-2 spike RBD and human ACE2. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Neerukonda, S.N.; Vassell, R.; Lusvarghi, S.; Wang, R.; Echegaray, F.; Bentley, L.; Eakin, A.E.; Erlandson, K.J.; Katzelnick, L.C.; Weiss, C.D.; et al. SARS-COV-2 Delta variant displays moderate resistance to neutralizing antibodies and spike protein properties of higher soluble ACE2 sensitivity, enhanced cleavage and fusogenic activity. Viruses 2021, 13, 2485. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, I.; Pravica, V.; Miljanovic, D.; Cupic, M. Immune Evasion of SARS-CoV-2 Emerging Variants: What Have We Learnt So Far? Viruses 2021, 13, 1192. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021, 27, 620–621. [Google Scholar] [CrossRef]
- Ewer, K.J.; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; Bittaye, M.; et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021, 27, 270–278. [Google Scholar] [CrossRef]
- Ravichandran, S.; Coyle, E.M.; Klenow, L.; Tang, J.; Grubbs, G.; Liu, S.; Wang, T.; Golding, H.; Khurana, S. Antibody signature induced by SARS-CoV-2 spike protein immunogens in rabbits. Sci. Transl. Med. 2020, 12, 550. [Google Scholar] [CrossRef]
- Gu, Y.; Cao, J.; Zhang, X.; Gao, H.; Wang, Y.; Wang, J.; He, J.; Jiang, X.; Zhang, J.; Shen, G.; et al. Receptome profiling identifies KREMEN1 and ASGR1 as alternative functional receptors of SARS-CoV-2. Cell Res. 2021, 1–14. [Google Scholar] [CrossRef]
- Chittum, J.E.; Sankaranarayanan, N.V.; O’Hara, C.P.; Desai, U.R. On the Selectivity of Heparan Sulfate Recognition by SARS-CoV-2 Spike Glycoprotein. ACS Med. Chem. Lett. 2021, 12, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mohammad, A.; Haq, I.; Nasar, M.; Ahmad, W.; Yousafi, Q.; Suleman, M.; Ahmad, S.; Albutti, A.; Khan, T.; et al. Structural-Dynamics and Binding Analysis of RBD from SARS-CoV-2 Variants of Concern (VOCs) and GRP78 Receptor Revealed Basis for Higher Infectivity. Microorganisms 2021, 9, 2331. [Google Scholar] [CrossRef] [PubMed]

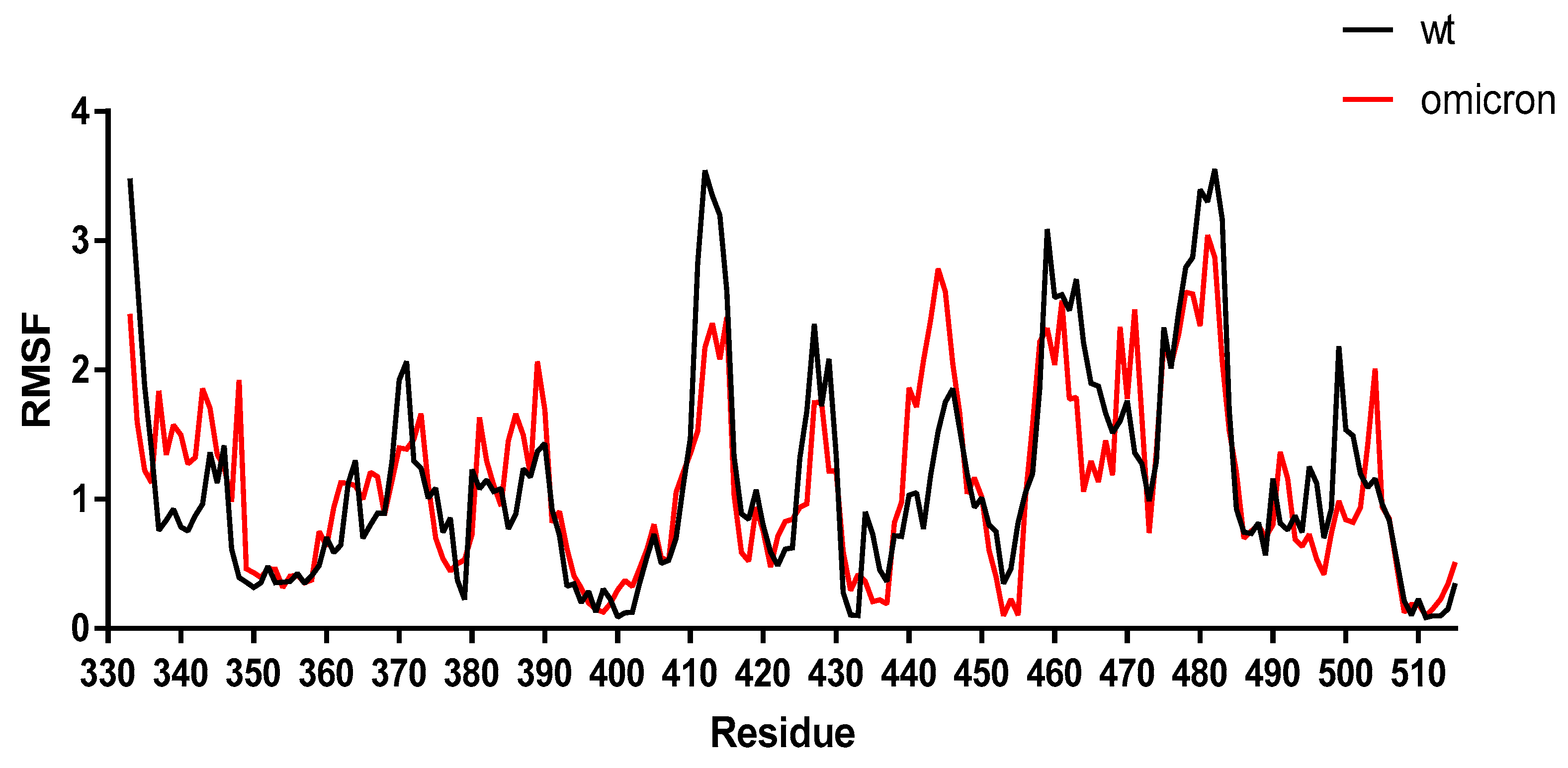

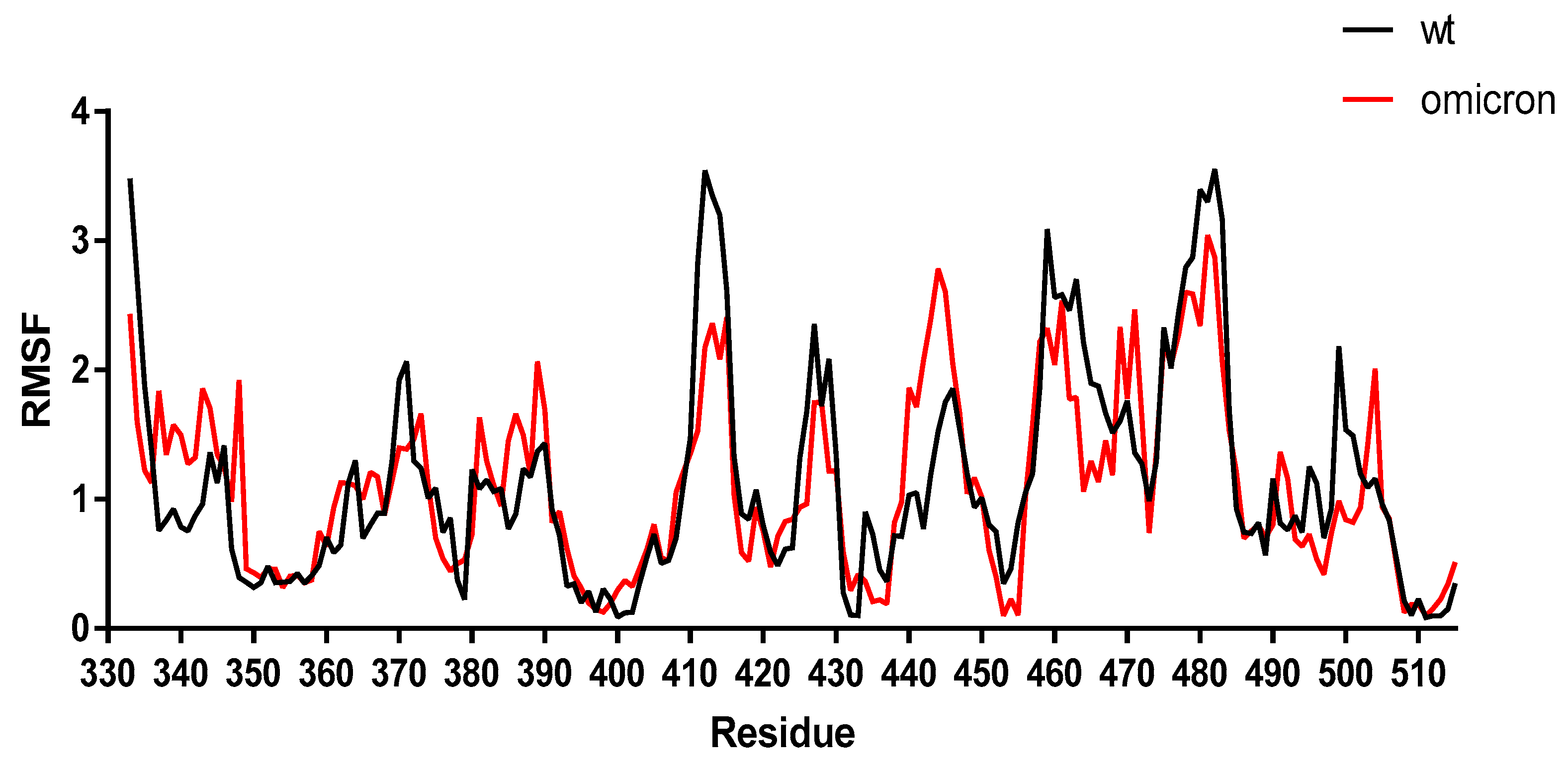
{kind=link}
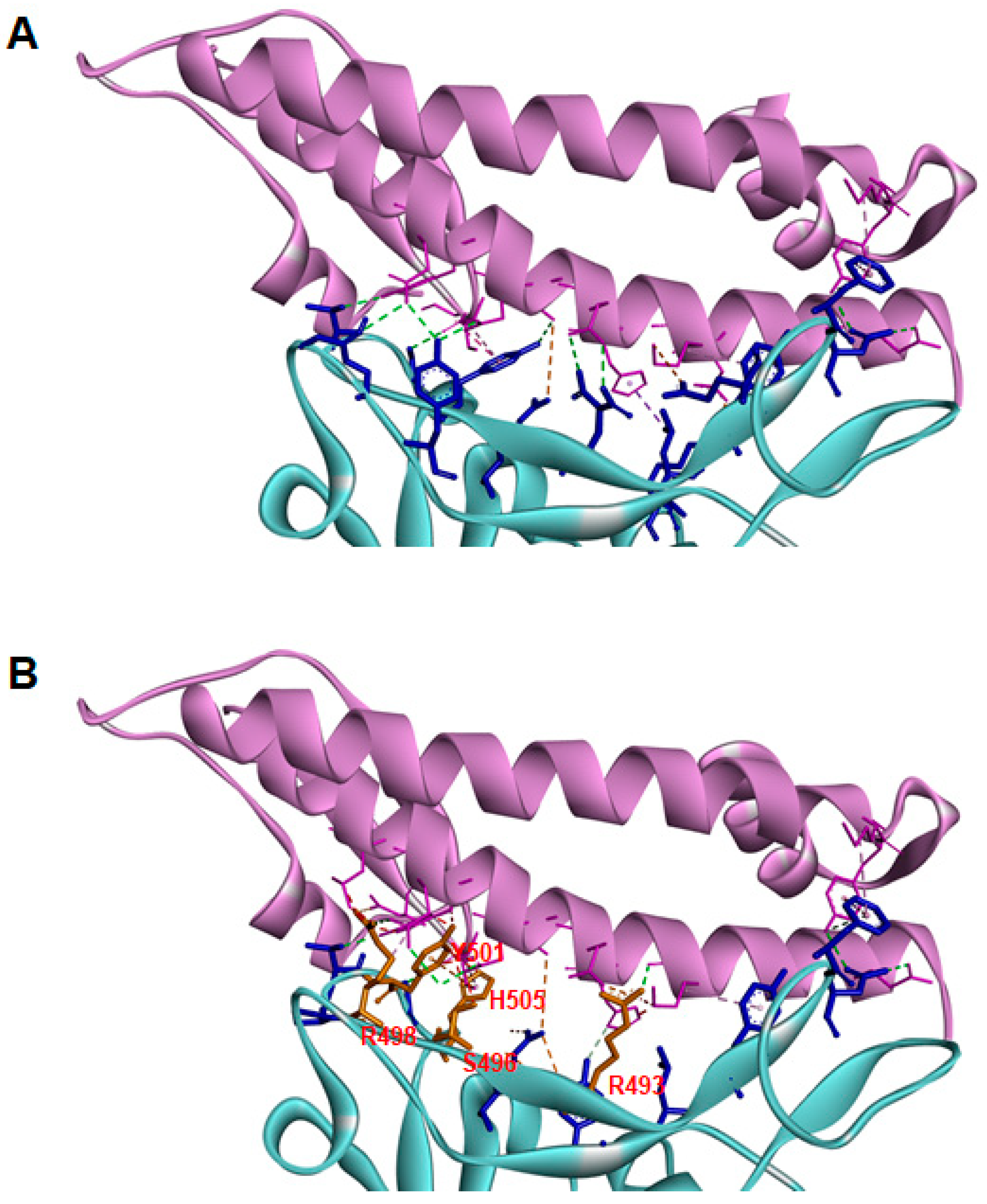
{kind=link}
| Residue | WT | Alpha | Beta | Gamma | Delta | Omicron | Mu |
|---|---|---|---|---|---|---|---|
| 339 | G | D | |||||
| 371 | S | L | |||||
| 373 | S | P | |||||
| 375 | S | F | |||||
| 417 | K | N | T | N | |||
| 440 | N | K | |||||
| 446 | G | S | |||||
| 452 | L | R | |||||
| 477 | S | N | |||||
| 478 | T | K | K | ||||
| 484 | E | K | K | A | K | ||
| 493 | Q | R | |||||
| 496 | G | S | |||||
| 498 | Q | R | |||||
| 501 | N | Y | Y | Y | Y | Y | |
| 505 | Y | H |
| Parameter | WT | Omicron |
|---|---|---|
| Binding energy (kcal/mol) | −11.3 | −12.6 |
| ICs charged-charged: | 4 | 12 |
| ICs charged-polar: | 10 | 8 |
| ICs charged-apolar: | 18 | 23 |
| ICs polar-polar: | 4 | 4 |
| ICs polar-apolar: | 22 | 18 |
| ICs apolar-apolar: | 10 | 13 |
| Total number of ICs | 68 | 78 |
| Parameter | WT | Omicron |
|---|---|---|
| HADDOCK score | −109.8 +/− 3.5 | −163.8 +/− 4.1 |
| van der Waals energy | −60.3 +/− 2.9 | −111.0 +/− 3.8 |
| Electrostatic energy | −148.9 +/− 44.9 | −382.8 +/− 42.1 |
| Desolvation energy | −34.2 +/− 9.2 | −13.9 +/− 5.2 |
| Buried Surface Area | 1778.5 +/− 96.8 | 2705.1 +/− 35.2 |
| Z-Score | −2.1 | −2.1 |
| ACE2 | WT Spike | Omicron Spike |
|---|---|---|
| S19 | A475 | A475 (2) |
| Q24 | A475, G476, S477, F486, N487, Y489 | A475, G476, A477, F486, N487, Y489 |
| T27 | F456, Y473, A475, Y489 | F456, Y473, A475, Y489 |
| P28 | N487, Y489 | N487, Y489 |
| D30 | K417, L455, F456 | N417, L455, F456, R493 |
| K31 | L455, F456, GLU484, Y489, F490, Q493 | L455, F456, Y489, F490, R493 |
| F32 | --- | R493 |
| H34 | K417, Y453, L455, Q493 | N417, Y453, L455, R493 |
| E35 | Q493, R403, Y505 | R493, R403, Y501, H505 |
| D38 | Y449, G496, Q498 | Y449, S496, R498, Y501 |
| F40 | --- | Y501 |
| Y41 | Q498, T500, N501 | R498, T500, Y501 |
| Q42 | V445, G446, G447, Y449, Q498 | V445, S446, G447, Y449, R498, Y501 |
| L45 | V445, Q498, T500, F486 | V445, R498, T500, F486 |
| M82 | F486 | F486 |
| Y83 | F486, N487, Y489 | F486, N487, Y489 |
| N330 | T500 | T500, T500 |
| L351 | --- | R498, Y501 |
| G352 | --- | Y501, H505 (2) |
| k353 | Y495, G496, F497, Q498, T500, N501, G502, Y505 | Y4895, S496, F497, R498, T500, Y501 |
| G354 | T500, N501, G502, V503, Y505 | T500, Y501, G502, V503, H505 |
| D355 | T500, N501, G502 | R498, T500, Y501, G502, H505 |
| R357 | T500 | R498, T500 |
| A386 | Y505 | --- |
| R393 | Y505 | --- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, J.T.; Jastrzebska, B.; Rangel, H.R. Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis. Pathogens 2022, 11, 45. https://doi.org/10.3390/pathogens11010045
Ortega JT, Jastrzebska B, Rangel HR. Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis. Pathogens. 2022; 11(1):45. https://doi.org/10.3390/pathogens11010045
Chicago/Turabian StyleOrtega, Joseph Thomas, Beata Jastrzebska, and Hector Rafael Rangel. 2022. "Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis" Pathogens 11, no. 1: 45. https://doi.org/10.3390/pathogens11010045
APA StyleOrtega, J. T., Jastrzebska, B., & Rangel, H. R. (2022). Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis. Pathogens, 11(1), 45. https://doi.org/10.3390/pathogens11010045