
Molecular Characterization of Bovine Leukemia Virus with the Evidence of a New Genotype Circulating in Cattle from Kazakhstan

 , and
, and
Abstract
:1. Introduction
2. Results
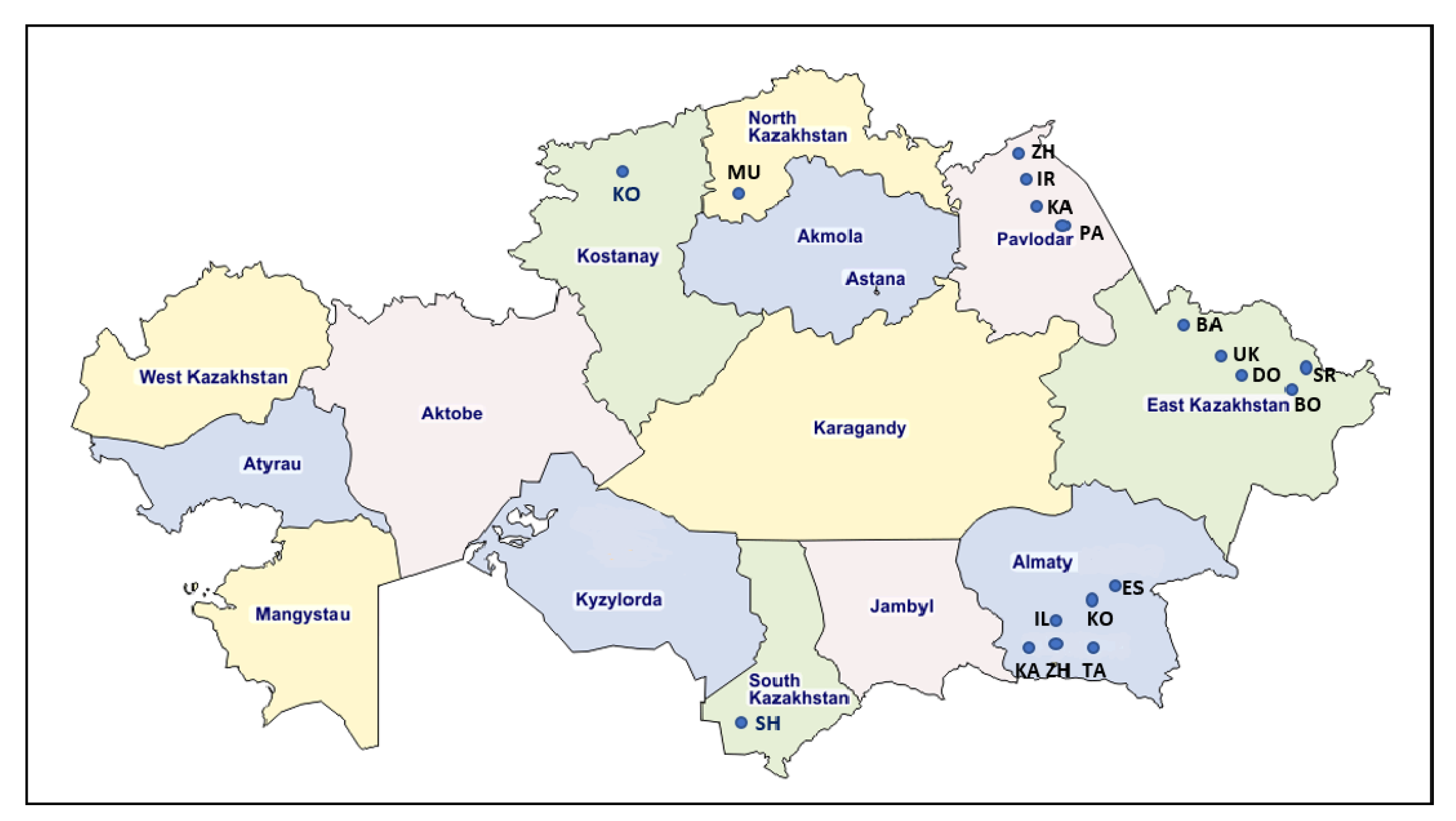
2.1. Detection of BLV-Infected Cattle by ELISA and qPCR
2.2. Estimation of BLV Proviral Load
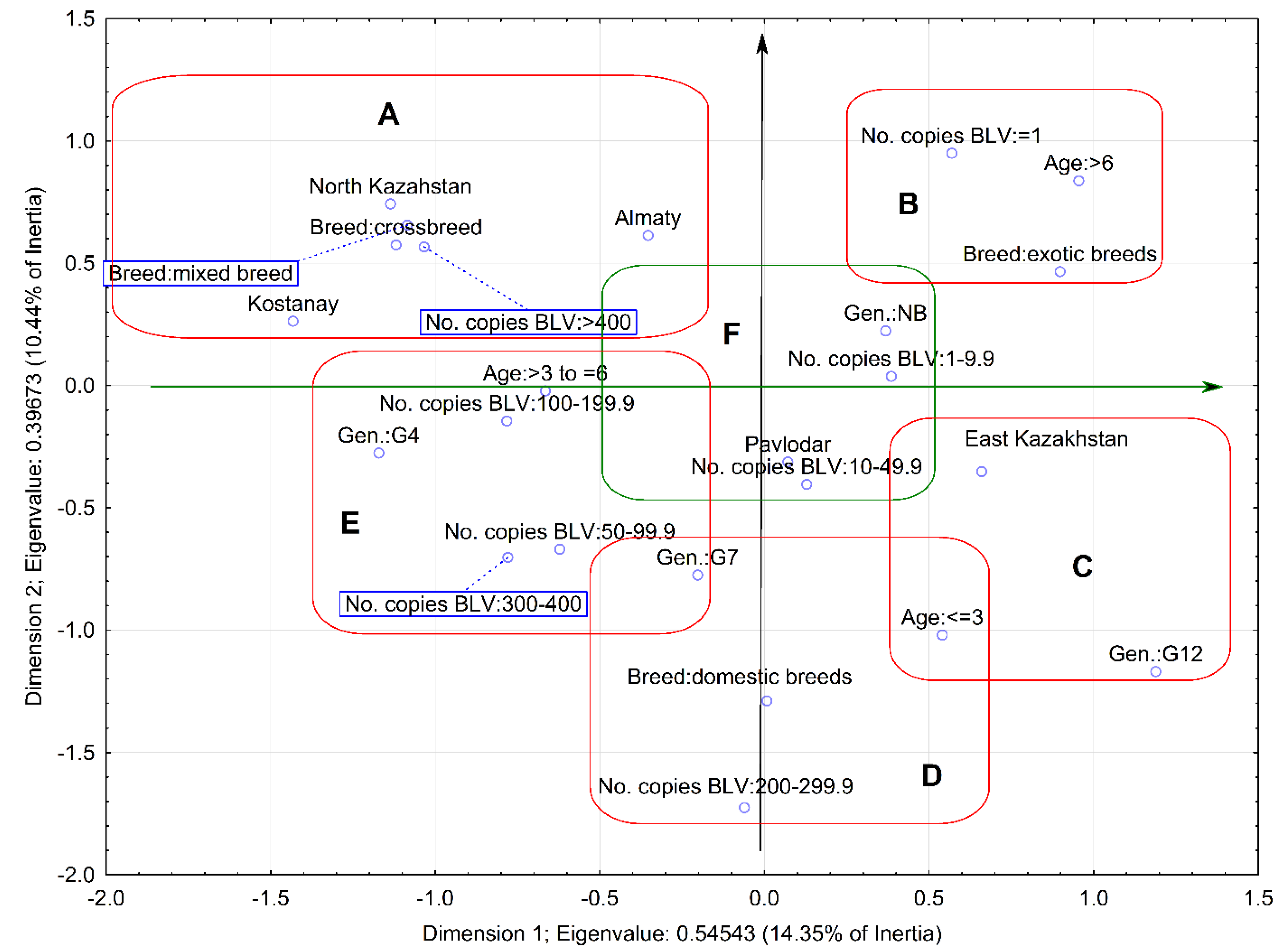
2.3. Relationship between Proviral Load and Variables
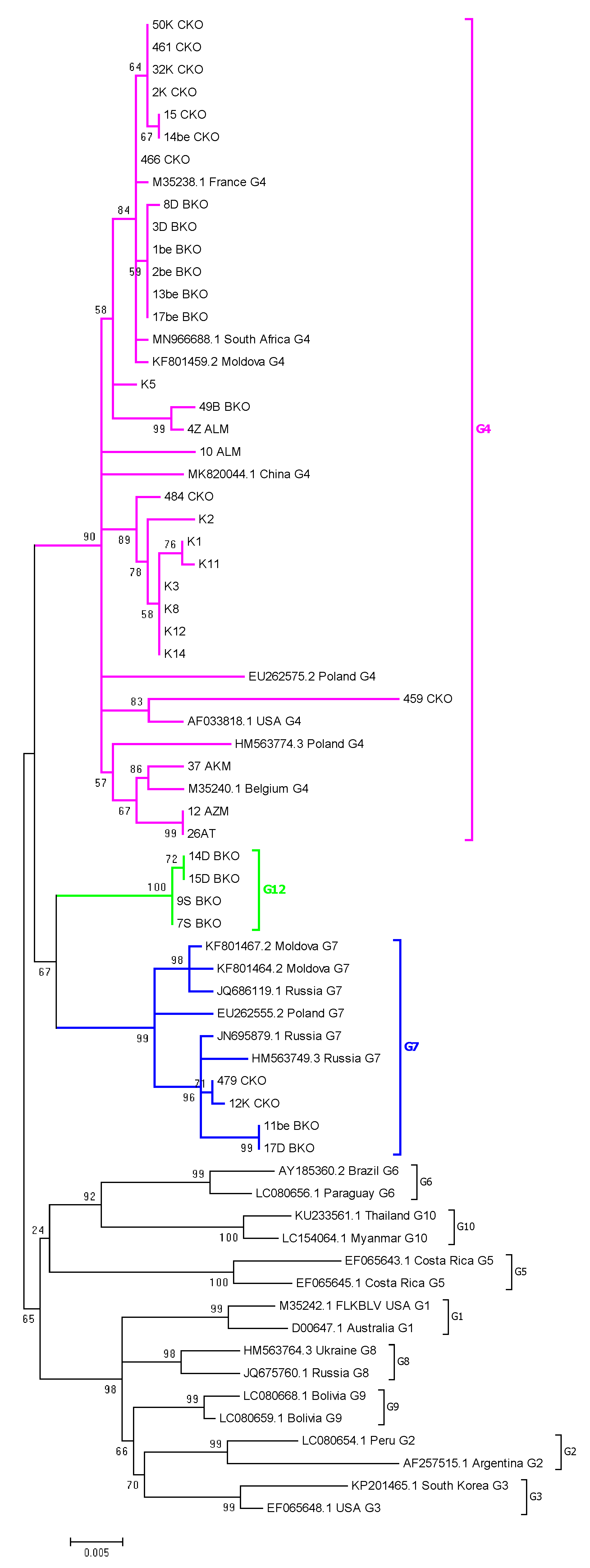
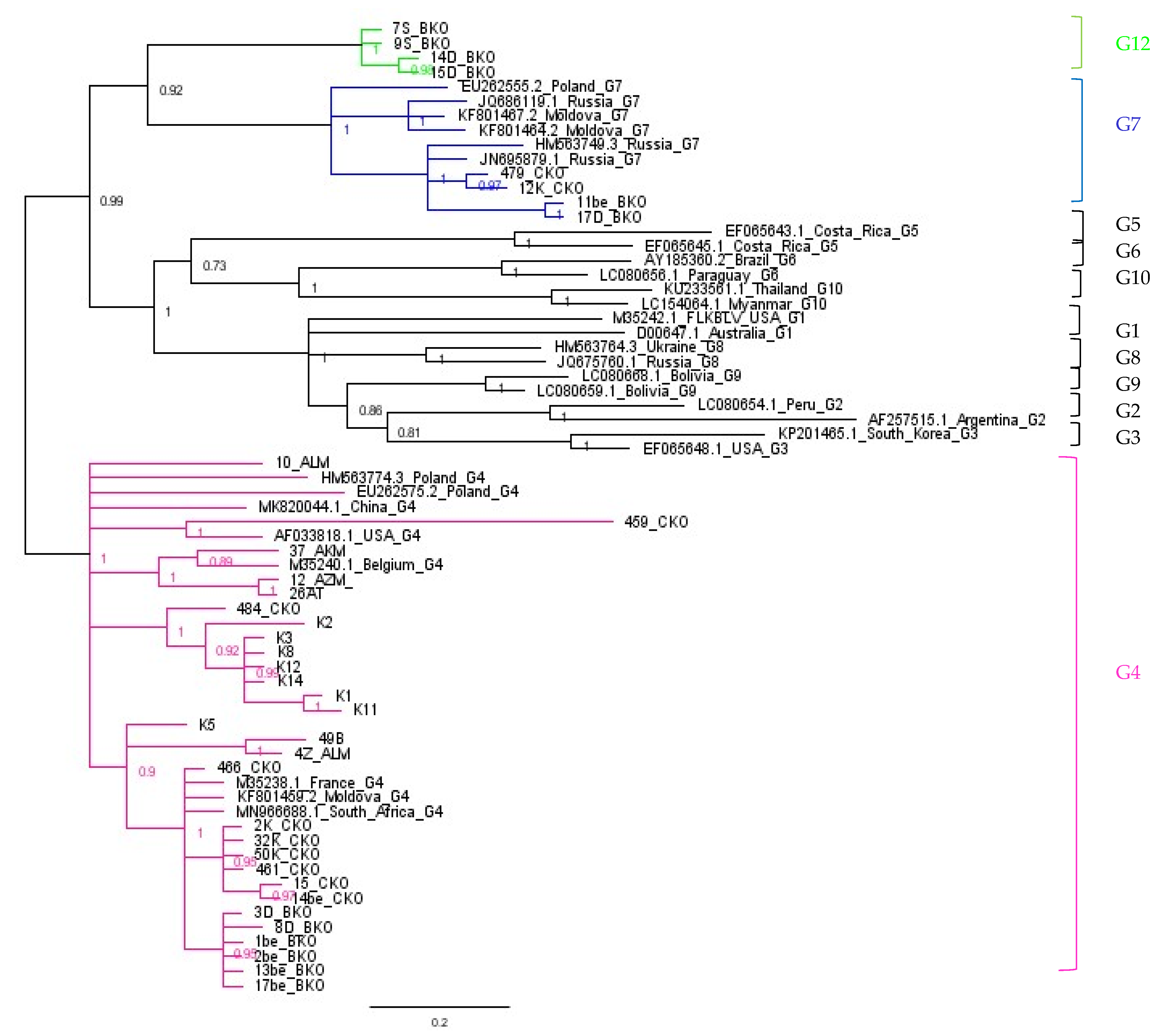
2.4. Phylogenetic Analysis Based on ML Method
2.5. Assignment of Genotype G12
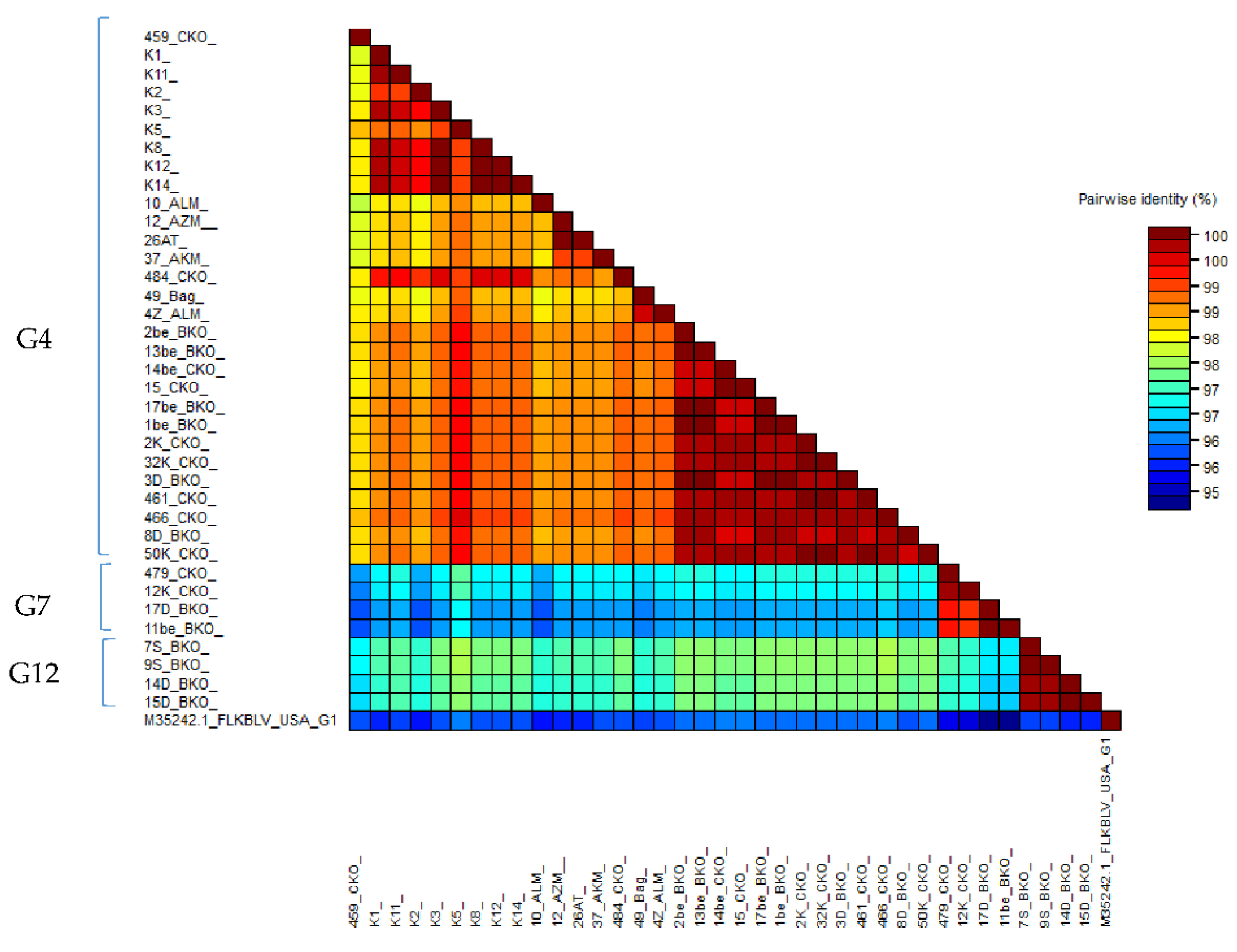
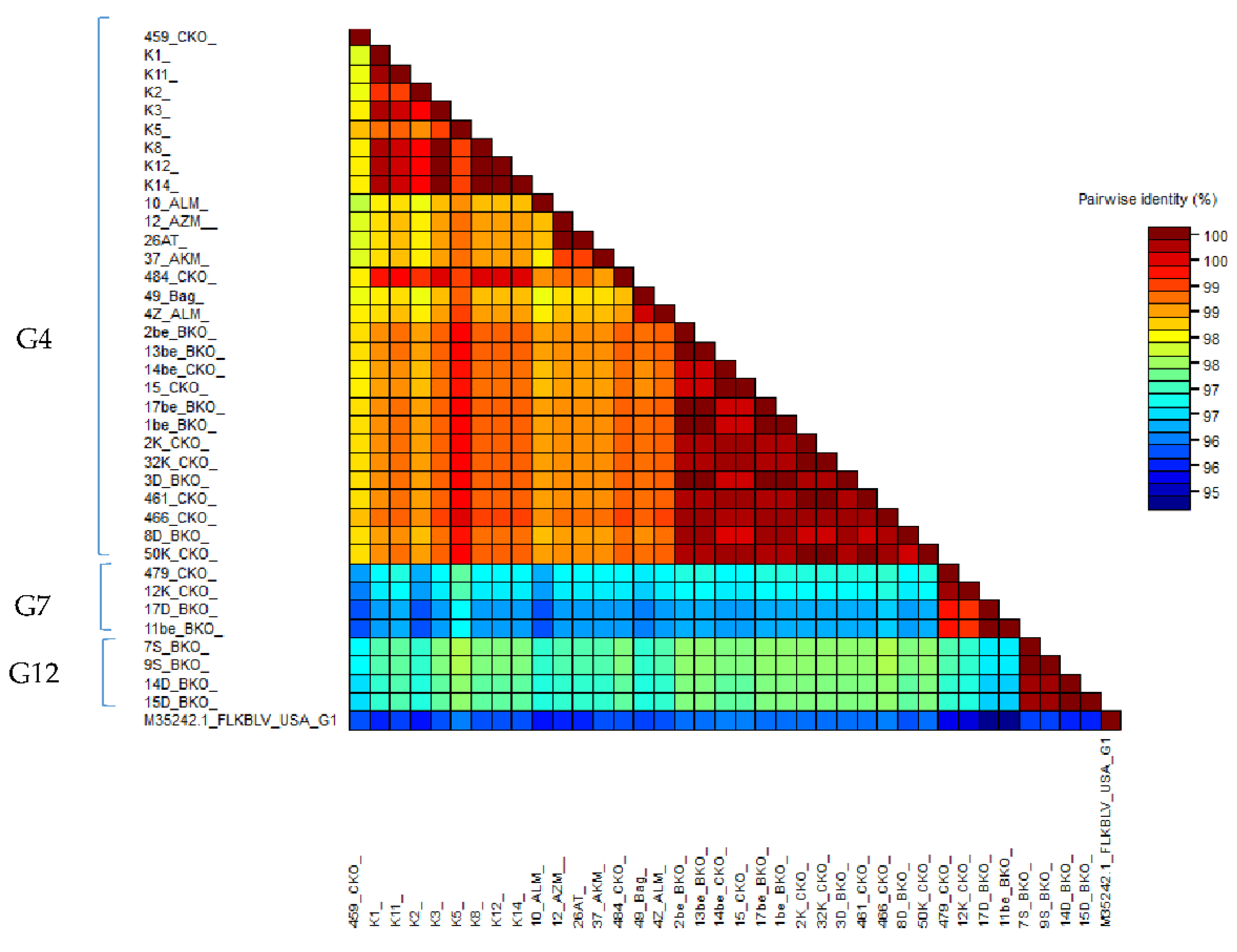
2.6. Comparison of Nucleotide Sequences Belonging to Genotype G4, G7 and G12
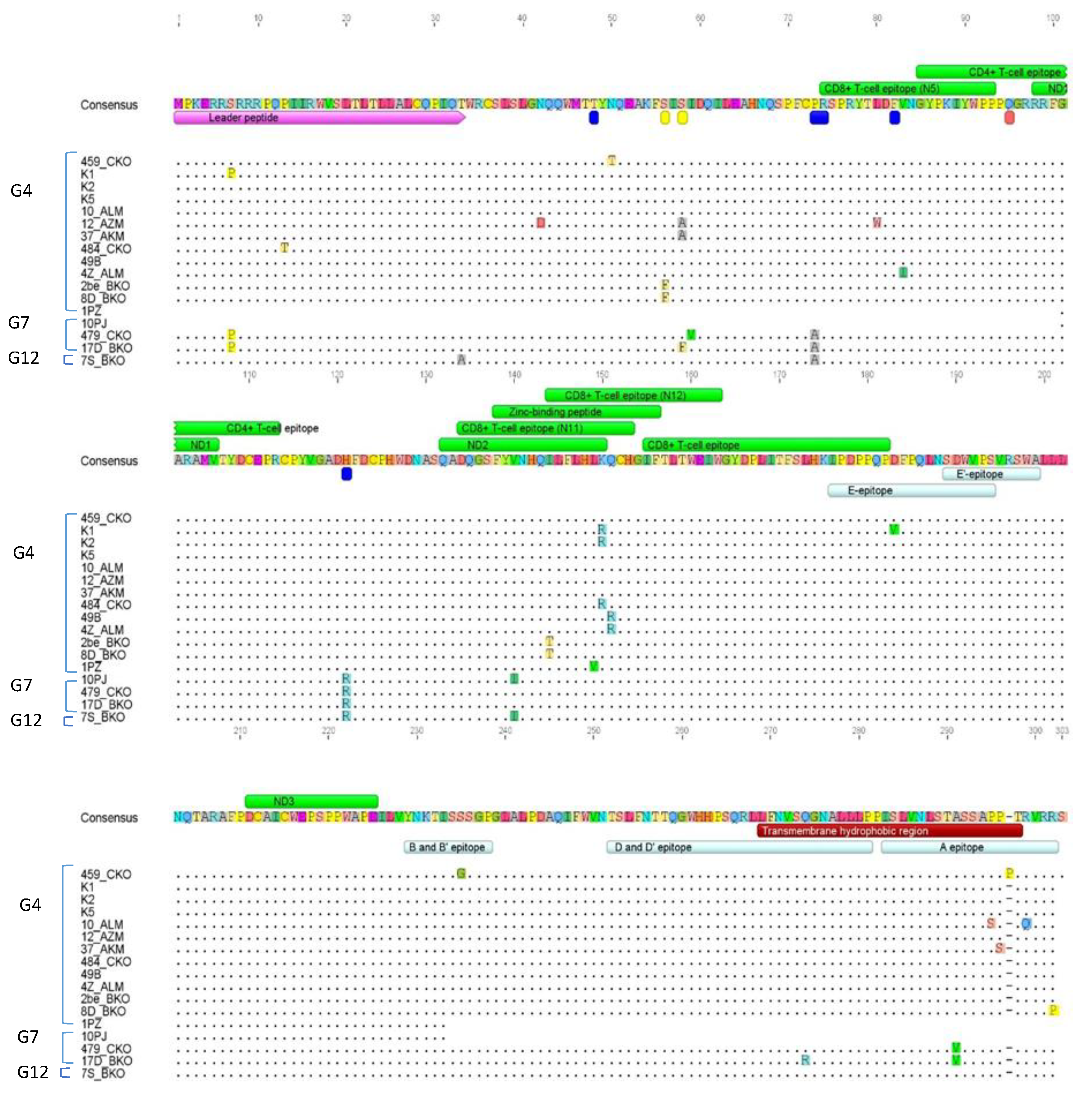
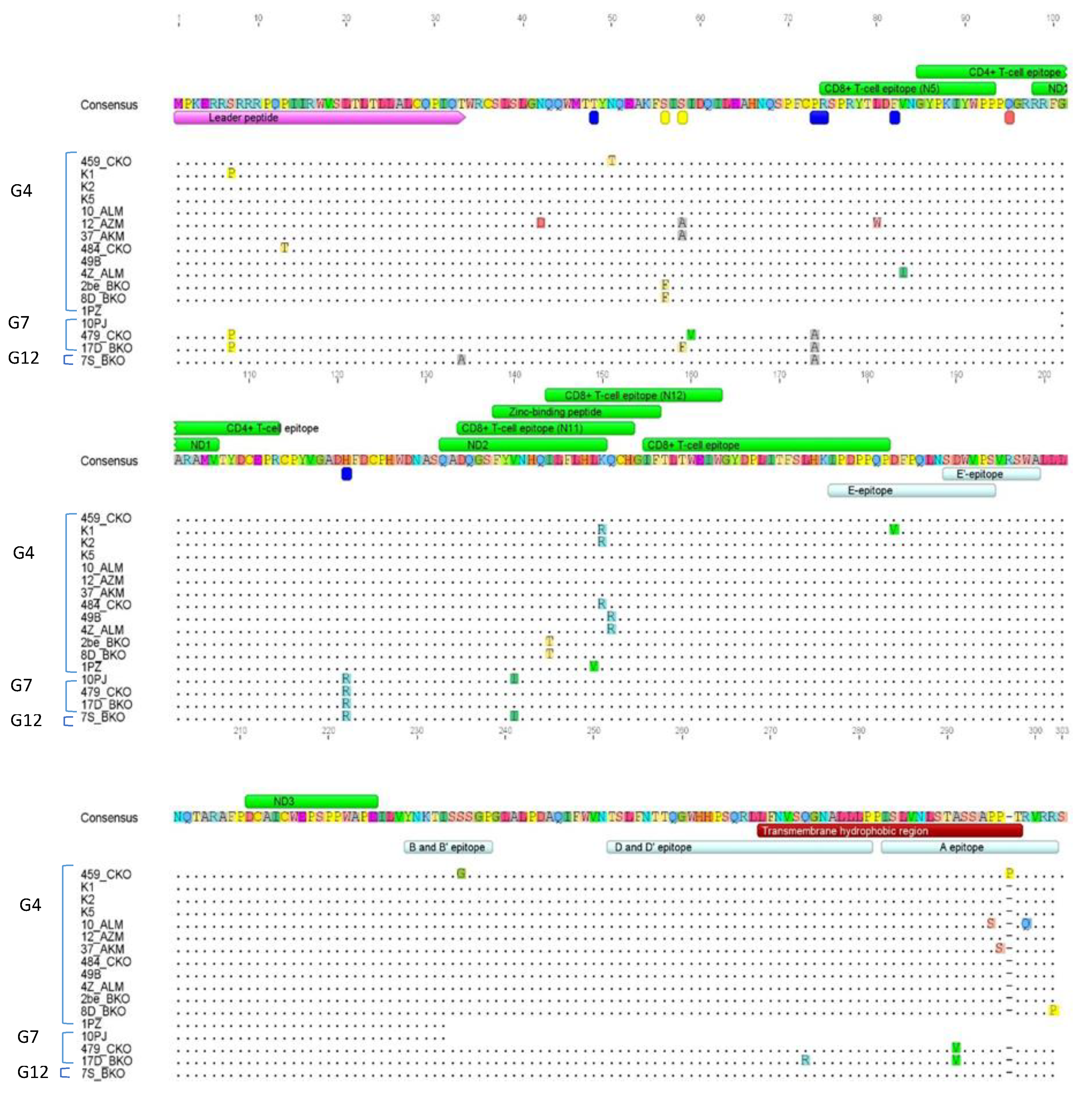
2.7. Amino Acid Sequence Analysis
3. Discussion
4. Materials and Methods
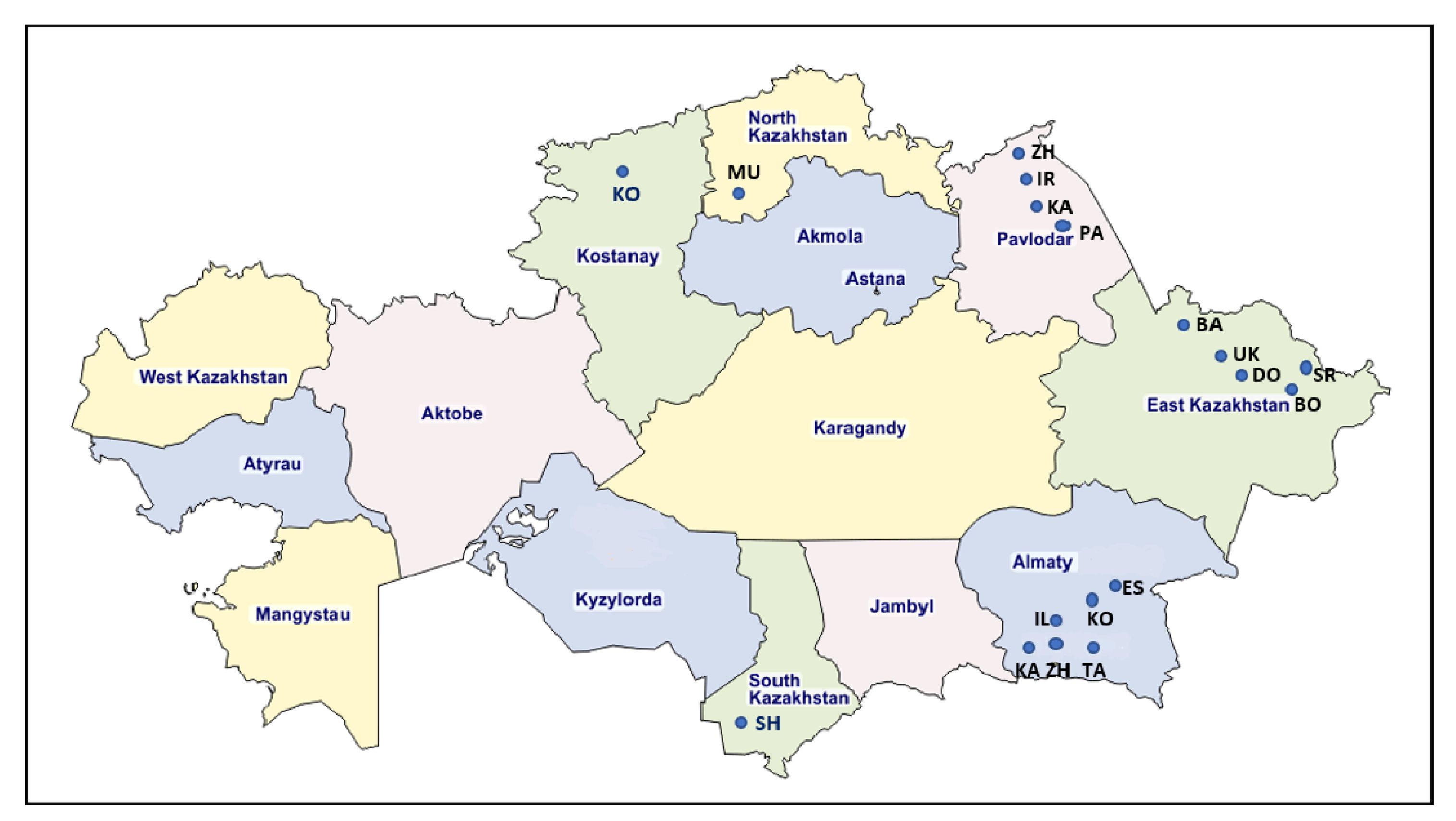
4.1. Sample Collection and DNA Extraction
4.2. Proviral Load Quantification
4.3. PCR Amplification of 993 bp and 444 bp Fragments of Env Gene
4.4. DNA Sequencing and Sequence Analysis
4.5. Multivariate Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hopkins, S.G.; DiGiacomo, R.F. Natural Transmission of Bovine Leukemia Virus in Dairy and Beef Cattle. Vet. Clin. N. Am. Food Anim. Pr. 1997, 13, 107–128. [Google Scholar] [CrossRef]
- Meas, S.; Usui, T.; Ohashi, K.; Sugimoto, C.; Onuma, M. Vertical transmission of bovine leukemia virus and bovine immunodeficiency virus in dairy cattle herds. Vet. Microbiol. 2002, 84, 275–282. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tsutsui, T.; Yamamoto, T.; Hayama, Y.; Kameyama, K.-I.; Konishi, M.; Murakami, K. Risk factors associated with within-herd transmission of bovine leukemia virus on dairy farms in Japan. BMC Vet. Res. 2010, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Panei, C.J.; Larsen, A.E.; Fuentealba, N.A.; Metz, G.E.; Echeverría, M.G.; Galosi, C.M.; Valera, A.R. Study of horn flies as vectors of bovine leukemia virus. Open Vet. J. 2019, 9, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Gillet, N.; Florins, A.; Boxus, M.; Burteau, C.; Nigro, A.; Vandermeers, F.; Balon, H.; Bouzar, A.-B.; Defoiche, J.; Burny, A.; et al. Mechanisms of leukemogenesis induced by bovine leukemia virus: Prospects for novel anti-retroviral therapies in human. Retrovirology 2007, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, P.C.; Ruggiero, V.J.; Hutchinson, H.C.; Droscha, C.J.; Norby, B.; Sporer, K.R.B.; Taxis, T.M. Current Developments in the Epidemiology and Control of Enzootic Bovine Leukosis as Caused by Bovine Leukemia Virus. Pathogens 2020, 9, 1058. [Google Scholar] [CrossRef]
- Ott, S.; Johnson, R.; Wells, S. Association between bovine-leukosis virus seroprevalence and herd-level productivity on US dairy farms. Prev. Vet. Med. 2003, 61, 249–262. [Google Scholar] [CrossRef]
- Erskine, R.J.; Bartlett, P.C.; Byrem, T.M.; Render, C.L.; Febvay, C.; Houseman, J.T. Herd-level determinants of bovine leukaemia virus prevalence in dairy farms. J. Dairy Res. 2012, 79, 445–450. [Google Scholar] [CrossRef]
- Bartlett, P.; Norby, B.; Byrem, T.; Parmelee, A.; Ledergerber, J.; Erskine, R. Bovine leukemia virus and cow longevity in Michigan dairy herds. J. Dairy Sci. 2013, 96, 1591–1597. [Google Scholar] [CrossRef] [Green Version]
- OIE (World Organization for Animal Health). Chapter 3.4.9 Enzootic Bovine Leucosis. In Terrestrial Animal Health Code; Office International des Epizooties: Paris, France, 2018; ISBN 978-92-95108-18-9. [Google Scholar]
- Kettmann, R.; Meunier-Rotival, M.; Cortadas, J.; Cuny, G.; Ghysdael, J.; Mammerickx, M.; Burny, A.; Bernardi, G. Integration of bovine leukemia virus DNA in the bovine genome. Proc. Natl. Acad. Sci. USA 1979, 76, 4822–4826. [Google Scholar] [CrossRef] [Green Version]
- Moe, K.K.; Polat, M.; Borjigin, L.; Matsuura, R.; Hein, S.T.; Moe, H.H.; Aida, Y. New evidence of bovine leukemia virus circulating in Myanmar cattle through epidemiological and molecular characterization. PLoS ONE 2020, 15, e0229126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somura, Y.; Sugiyama, E.; Fujikawa, H.; Murakami, K. Comparison of the copy numbers of bovine leukemia virus in the lymph nodes of cattle with enzootic bovine leukosis and cattle with latent infection. Arch. Virol. 2014, 159, 2693–2697. [Google Scholar] [CrossRef] [PubMed]
- Ohno, A.; Takeshima, S.; Matsumoto, Y.; Aida, Y. Risk factors associated with increased bovine leukemia virus proviral load in infected cattle in Japan from 2012 to 2014. Virus Res. 2015, 210, 283–290. [Google Scholar] [CrossRef]
- Lo, C.-W.; Takeshima, S.-N.; Okada, K.; Saitou, E.; Fujita, T.; Matsumoto, Y.; Wada, S.; Inoko, H.; Aida, Y. Association of Bovine Leukemia Virus-Induced Lymphoma with BoLA-DRB3 Polymorphisms at DNA, Amino Acid, and Binding Pocket Property Levels. Pathogens 2021, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Hamada, R.; Metwally, S.; Polat, M.; Borjigin, L.; Ali, A.O.; Abdel-Hady, A.A.A.; Mohamed, A.E.A.; Wada, S.; Aida, Y. Detection and Molecular Characterization of Bovine Leukemia Virus in Egyptian Dairy Cattle. Front. Vet. Sci. 2020, 7, 608. [Google Scholar] [CrossRef] [PubMed]
- Johnston, E.R.; Albritton, L.M.; Radke, K. Envelope Proteins Containing Single Amino Acid Substitutions Support a Structural Model of the Receptor-Binding Domain of Bovine Leukemia Virus Surface Protein. J. Virol. 2002, 76, 10861–10872. [Google Scholar] [CrossRef] [Green Version]
- Bruck, C.; Portetelle, D.; Burny, A.; Zavada, J. Topographical analysis by monoclonal antibodies of BLV-gp51 epitopes involved in viral functions. Virology 1982, 122, 353–362. [Google Scholar] [CrossRef]
- Callebaut, I.; Vonèche, V.; Mager, A.; Fumière, O.; Krchnak, V.; Merza, M.; Zavada, J.; Mammerickx, M.; Burny, A.; Portetelle, D. Mapping of B-neutralizing and T-helper cell epitopes on the bovine leukemia virus external glycoprotein gp51. J. Virol. 1993, 67, 5321–5327. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Takeshima, S.; Isogai, E.; Kohara, J.; Aida, Y. Novel CD8+ cytotoxic T cell epitopes in bovine leukemia virus with cattle. Vaccine 2015, 33, 7194–7202. [Google Scholar] [CrossRef] [Green Version]
- Polat, M.; Takeshima, S.-N.; Aida, Y. Epidemiology and genetic diversity of bovine leukemia virus. Virol. J. 2017, 14, 209. [Google Scholar] [CrossRef]
- Yu, C.; Wang, X.; Zhou, Y.; Wang, Y.; Zhang, X.; Zheng, Y. Genotyping bovine leukemia virus in dairy cattle of Heilongjiang, northeastern China. BMC Vet. Res. 2019, 15, 179. [Google Scholar] [CrossRef]
- De Brogniez, A.; Bouzar, A.B.; Jacques, J.-R.; Cosse, J.-P.; Gillet, N.; Callebaut, I.; Reichert, M.; Willems, L. Mutation of a Single Envelope N-Linked Glycosylation Site Enhances the Pathogenicity of Bovine Leukemia Virus. J. Virol. 2015, 89, 8945–8956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brogniez, A.; Mast, J.; Willems, L. Determinants of the Bovine Leukemia Virus Envelope Glycoproteins Involved in Infectivity, Replication and Pathogenesis. Viruses 2016, 8, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agency of Statistics of the Republic of Kazakhstan database. Available online: https://stat.gov.kz/official/industry/14/statistic/8 (accessed on 25 November 2021).
- Bakhtakhunov, Y.K.; Baramova, S.A.; Aitlesova, R.B. Leukemia of cattle and measures to combat it. Vestn. Agric. Sci. Kazakhstan 2011, 12, 25–55. [Google Scholar]
- Mamanova, S.B.; Kalisynov, B.S.; Saduakasova, M.A.; Bashenova, E.E.; Maukish, A.N.A. Analysis of the epizootic situation on bovine leukemia for 2015–2019 in the East Kazakhstan region. Collect. KazSRVI 2020, 66, 60–64. [Google Scholar]
- Safronova, O.S.; Lykasova, I.A. Use of method PCR in veterinary-sanitary analysis on a leukaemia (BLV). Sci. Technol. Educ. Sci. 2019, 3, 94–99. [Google Scholar]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Le, D.T.; Yamashita-Kawanishi, N.; Okamoto, M.; Nguyen, S.V.; Nguyen, N.H.; Sugiura, K.; Miura, T.; Haga, T. Detection and genotyping of bovine leukemia virus (BLV) in Vietnamese cattle. J. Vet. Med. Sci. 2020, 82, 1042–1050. [Google Scholar] [CrossRef]
- Zinovieva, N.; Gladyr, E.; Vinogradova, I.; Mikhailova, M.; Molofeeva, L.E.L. Prevalence of bovine leukemia virus in black and with cows with the different level of milk productive traits. Selskokhozyaistvennaya Biol. 2012, 6, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Ochirkhuu, N.; Konnai, S.; Odbileg, R.; Nishimori, A.; Okagawa, T.; Murata, S.; Ohashi, K. Detection of bovine leukemia virus and identification of its genotype in Mongolian cattle. Arch. Virol. 2016, 161, 985–991. [Google Scholar] [CrossRef]
- Yang, Y.; Chu, S.; Shang, S.; Yang, Z.; Wang, C. Short communication: Genotyping and single nucleotide polymorphism analysis of bovine leukemia virus in Chinese dairy cattle. J. Dairy Sci. 2019, 102, 3469–3473. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-W.; Borjigin, L.; Saito, S.; Fukunaga, K.; Saitou, E.; Okazaki, K.; Mizutani, T.; Wada, S.; Takeshima, S.-N.; Aida, Y. BoLA-DRB3 Polymorphism is Associated with Differential Susceptibility to Bovine Leukemia Virus-Induced Lymphoma and Proviral Load. Viruses 2020, 12, 352. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, V.; Norby, B.; Benitez, O.; Hutchinson, H.; Sporer, K.; Droscha, C.; Swenson, C.; Bartlett, P. Controlling bovine leukemia virus in dairy herds by identifying and removing cows with the highest proviral load and lymphocyte counts. J. Dairy Sci. 2019, 102, 9165–9175. [Google Scholar] [CrossRef] [Green Version]
- Juliarena, M.A.; Barrios, C.N.; Ceriani, M.C.; Esteban, E.N. Hot topic: Bovine leukemia virus (BLV)-infected cows with low proviral load are not a source of infection for BLV-free cattle. J. Dairy Sci. 2016, 99, 4586–4589. [Google Scholar] [CrossRef] [Green Version]
- Mekata, H.; Yamamoto, M.; Hayasi, T.; Kirino, Y.; Sekiguchi, S.; Konnai, S.; Horii, Y.N.J. Cattle with a low bovine leukemia virus proviral load are rarely an infectious source. Jpn. J. Vet. Res. 2018, 66, 157–163. [Google Scholar] [CrossRef]
- Hutchinson, H.C.; Norby, B.; Droscha, C.J.; Sordillo, L.M.; Coussens, P.M.; Bartlett, P.C. Bovine leukemia virus detection and dynamics following experimental inoculation. Res. Vet. Sci. 2020, 133, 269–275. [Google Scholar] [CrossRef]
- Mirsky, M.L.; Olmstead, C.A.; Da, Y.; Lewin, H.A. The prevalence of proviral bovine leukemia virus in peripheral blood mononuclear cells at two subclinical stages of infection. J. Virol. 1996, 70, 2178–2183. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, E.; Rosa, G.J.M.; Weigel, K.; Byrem, T. Genetic analysis of leukosis incidence in United States Holstein and Jersey populations. J. Dairy Sci. 2013, 96, 6022–6029. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Mekata, H.; Sekiguchi, S.; Kirino, Y.; Mitoma, S.; Honkawa, K.; Horii, Y.; Norimine, J. Cattle with the BoLA class II DRB3*0902 allele have significantly lower bovine leukemia proviral loads. J. Veter. Med. Sci. 2017, 79, 1552–1555. [Google Scholar] [CrossRef] [Green Version]
- Miyasaka, T.; Takeshima, S.-N.; Jimba, M.; Matsumoto, Y.; Kobayashi, N.; Matsuhashi, T.; Sentsui, H.; Aida, Y. Identification of bovine leukocyte antigen class II haplotypes associated with variations in bovine leukemia virus proviral load in Japanese Black cattle. Tissue Antigens 2013, 81, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kobayashi, S.; Konishi, M.; Kameyama, K.-I.; Yamamoto, T.; Tsutsui, T. The recent prevalence of bovine leukemia virus (BLV) infection among Japanese cattle. Vet. Microbiol. 2011, 148, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Şevik, M.; Avcı, O.; Ince, O.B. An 8-year longitudinal sero-epidemiological study of bovine leukaemia virus (BLV) infection in dairy cattle in Turkey and analysis of risk factors associated with BLV seropositivity. Trop. Anim. Health Prod. 2015, 47, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Morovati, H.; Shirvani, E.; Noaman, V.; Lotfi, M.; Kamalzadeh, M.; Hatami, A.; Bahreyari, M.; Shahramyar, Z.; Morovati, M.H.; Azimi, M.; et al. Seroprevalence of bovine leukemia virus (BLV) infection in dairy cattle in Isfahan Province, Iran. Trop. Anim. Heal. Prod. 2011, 44, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Sultanov, A.A.; Shmanov, G.S.; Kutumbetov, L.B.; Zhusupov, G.K. Recommendations for Maintaining Well-Being on Bluetongue and Schmallenberg Diseases in the Republic of Kazakhstan; KazSRVI: Almaty, Kazakhstan, 2016. [Google Scholar]
- Karymsakov, T.N.; Baimukanov, D.A. Milk productivity of cows of the active part of the Simmental breed population. Agrar. Sci. 2020, 39–41. [Google Scholar] [CrossRef]
- Rola-Łuszczak, M.; Pluta, A.; Olech, M.; Donnik, I.; Petropavlovskiy, M.; Gerilovych, A.; Vinogradova, I.; Choudhury, B.; Kuźmak, J. The Molecular Characterization of Bovine Leukaemia Virus Isolates from Eastern Europe and Siberia and Its Impact on Phylogeny. PLoS ONE 2013, 8, e58705. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, L.; Dong, M.; Huang, W.; Hao, X.; Peng, Y.; Gong, Z.; Qin, A.; Shang, S.; Yang, Z. Molecular characterization of bovine leukemia virus reveals existence of genotype 4 in Chinese dairy cattle. Virol. J. 2019, 16, 108. [Google Scholar] [CrossRef] [Green Version]
- Casal, J.; Learte, P.; Torre, E. A path model of factors influencing bovine leukemia virus transmission between cattle herds. Prev. Vet. Med. 1990, 10, 47–61. [Google Scholar] [CrossRef]
- Mekata, H.; Sekiguchi, S.; Konnai, S.; Kirino, Y.; Horii, Y.; Norimine, J. Horizontal transmission and phylogenetic analysis of bovine leukemia virus in two districts of Miyazaki, Japan. J. Vet. Med. Sci. 2015, 77, 1115–1120. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Buehring, G.C. Natural genetic variations in bovine leukemia virus envelope gene: Possible effects of selection and escape. Virology 2007, 366, 150–165. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, Y.; Baloch, A.R.; Pan, Y.; Xu, F.; Tian, L.; Zeng, Q. Molecular epidemiology and characterization of bovine leukemia virus in domestic yaks (Bos grunniens) on the Qinghai-Tibet Plateau, China. Arch. Virol. 2018, 163, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Rola-Łuszczak, M.; Kubis, P.; Balov, S.; Moskalik, R.; Choudhury, B.; Kuźmak, J. Molecular characterization of bovine leukemia virus from Moldovan dairy cattle. Arch. Virol. 2017, 162, 1563–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, S.M.; Golemba, M.D.; Campos, R.H.; Trono, K.; Jones, L.R. Bovine leukemia virus can be classified into seven genotypes: Evidence for the existence of two novel clades. J. Gen. Virol. 2009, 90, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Bazzucchi, M.; Iscaro, C.; Casciari, C.; Giammarioli, M.; Feliziani, F. Molecular characterization of Italian bovine leukemia virus isolates reveals the presence of distinct phylogenetic clusters. Arch. Virol. 2019, 164, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Polat, M.; Ohno, A.; Takeshima, S.; Kim, J.; Kikuya, M.; Matsumoto, Y.; Mingala, C.N.; Onuma, M.; Aida, Y. Detection and molecular characterization of bovine leukemia virus in Philippine cattle. Arch. Virol. 2014, 160, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Sood, R. Molecular Characterization of Bovine Leukaemia Virus (BLV) Strains Reveals Existence of Genotype 6 in Cattle in India with evidence of a new subgenotype. Transbound. Emerg. Dis. 2018, 65, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Rola-Łuszczak, M.; Finnegan, C.; Olech, M.; Choudhury, B.; Kuźmak, J. Development of an improved real time PCR for the detection of bovine leukaemia provirus nucleic acid and its use in the clarification of inconclusive serological test results. J. Virol. Methods 2013, 189, 258–264. [Google Scholar] [CrossRef]
- Pluta, A.; Blazhko, N.; Ngirande, C.; Joris, T.; Willems, L.; Kuźmak, J. Analysis of Nucleotide Sequence of Tax, miRNA and LTR of Bovine Leukemia Virus in Cattle with Different Levels of Persistent Lymphocytosis in Russia. Pathogens 2021, 10, 246. [Google Scholar] [CrossRef]
- Rulka, J.; Kubis, P.; Deren, W.B.E. Evaluation of the nested-PCR method for the diagnosis of bovine leukaemia virus (BLV) infection. Bull. Vet. Inst. Pulawy 2001, 45, 11–19. [Google Scholar]
- Drummond, A.J.; Ashton, B.; Buxton, S.; Cheung, M.; Cooper, A.; Duran, C.; Field, M.; Heled, J.; Kearse, M.; Markovitz, S.; et al. Geneious v5.5. 2011. Available online: http://www.geneious.com (accessed on 25 November 2021).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Greenacre, M.H.T. The geometric interpretation of correspondence analysis. J. Am. Stat. Assoc. 1987, 82, 437–447. [Google Scholar] [CrossRef]
- Benzecri, J.-P. Correspondence analysis. Cahiers de l’Analyse des Donnees (Data Analysis Book); Dunod: Paris, France, 1973. [Google Scholar]
- Lebart, L.; Morineau, A.; Tabard, N. Techniques de la Description Statistique: Methodes et Logiciels Pour IAnalyse des Grands Tableaux; Dunod: Paris, France, 1977. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Farm ID | Breed | Age (y.m) | Seropositive No./ Tested No. (Positive %) | Positive No./Tested No. by qPCR | Number of Provirus Copies Per 1000 Cells (Range, Median Value ) | Number of Sequences 903 bp | Number of Sequences 444 bp | Genotypes |
|---|---|---|---|---|---|---|---|---|---|
| Kostanay | KO | Black and motley | 3.6–5.0 | 14/30 (46.6) | 14/14 | 1.0–542.8 184.8 | 8 | 0 | G4 (8) |
| North Kazakhstan | MU | Black and motley | 5.0–11.0 | 43/88 (48.8) | 17/17 | 1.0–606.0 225.5 | 11 | 0 | G4 (9) G7 (2) |
| Pavlodar | IR | Simmental | 4.0–5.0 | 2/64 (3.1) | 2/2 | 1.0–2.3 1.0 | 0 | 1 | G7 (1) |
| ZH | Kazakh—white-headed | 4.0 | 1/125 (0.8) | 1/1 | 1.2 | 0 | 1 | G4 (1) | |
| PA | Simmental | 4.0–5.0 | 2/120 (1.7) | 2/2 | 1.0–45.1 22.8 | 0 | 1 | G7 (1) | |
| KA | Kazakh—white-headed | 5.0 | 1/65 (1.5) | 1/1 | 40.0 | 0 | 1 | G7 (1) | |
| East Kazakhstan | BA | Simmental | 1.6 | 20/50 (40.0) | 12/16 | 1.0–66.2 17.6 | 1 | 0 | G4 (1) |
| UK | Simmental | 5.0–10.5 | 27/50 (54.0) | 16/20 | 1.0–348.1 87.3 | 0 | 0 | 0 | |
| SR | Simmental | 6.0–14.1 | 19/30 (63.3) | 12/19 | 1.0–18.4 5.2 | 2 | 0 | G12 (2) | |
| DO | Alatau/Local, without breed | 2.0–3.9 | 15/25 (60.0) | 13/14 | 11.5–367.7 91.3 | 5 | 0 | G4 (2) G7 (1) G12 (2) | |
| BO | Local, without breed | 2.1–4.9 | 21/25 (84.0) | 14/19 | 107.3–484.7 114.3 | 5 | 0 | G4 (4) G7 (1) | |
| Almaty | ES | Local, without breed | 6.0–7.4 | 2/40 (5.0) | 0/2 | 0 | 0 | 0 | 0 |
| ZH | Santa Gertrude | 4.0–6.0 | 28/50 (56.0) | 10/23 | 1.0–481.9 182.7 | 3 | 0 | G4 (3) | |
| TA | Holstein/ Black and motley | 4.0 | 5/30 (16.7) | 2/3 | 28.0–879.0 425.2 | 1 | 0 | G4 (1) | |
| KA | Local, without breed | 6.0 | 1/40 (2.5) | 1/1 | 182.9 | 0 | 0 | 0 | |
| KO | Local, without breed | 5.0 | 4/40 (10.0) | 1/2 | 120.7 | 1 | 0 | G4 (1) | |
| IL | Black and motley | 5.0 | 3/20 (15.0) | 1/3 | 1.0 | 0 | 0 | 0 | |
| South Kazakhstan | SH | Local, without breed | 27/40 (67.5) | 0/27 | 0 | 0 | 0 | 0 | |
| Total | 18 | 228/962 (23.7) | 119/186 (63.9) | 37 | 4 | G4 (30) G7 (7) G12 (4) |
| G1 | G2 | G3 | G4 | G5 | G6 | G7 | G8 | G9 | G10 | G12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| G1 | 0.008 (±0.001) | ||||||||||
| G2 | 0.030 ± 0.005 | 0.009 (±0.002) | |||||||||
| G3 | 0.030 ± 0.004 | 0.028 ± 0.004 | 0.010 (±0.002) | ||||||||
| G4 | 0.037 ± 0.005 | 0.035 ± 0.005 | 0.039 ± 0.005 | 0.011 (±0.002) | |||||||
| G5 | 0.046 ± 0.006 | 0.047 ± 0.006 | 0.050 ± 0.006 | 0.037 ± 0.005 | 0.012 (±0.002) | ||||||
| G6 | 0.041 ± 0.005 | 0.042 ± 0.005 | 0.042 ± 0.005 | 0.035 ± 0.005 | 0.045 ± 0.006 | 0.014 (±0.002) | |||||
| G7 | 0.040±0.005 | 0.039 ± 0.005 | 0.042 ± 0.006 | 0.029 ± 0.004 | 0.045 ± 0.006 | 0.041 ± 0.005 | 0.009 (±0.002) | ||||
| G8 | 0.023 ± 0.004 | 0.026 ± 0.004 | 0.027 ± 0.004 | 0.033 ± 0.005 | 0.045 ± 0.006 | 0.034 ± 0.005 | 0.036 ± 0.005 | 0.009 (±0.002) | |||
| G9 | 0.023 ± 0.004 | 0.022 ± 0.004 | 0.022 ± 0.004 | 0.031 ± 0.005 | 0.043 ± 0.006 | 0.036 ± 0.005 | 0.035 ± 0.005 | 0.019 ± 0.004 | 0.001 (±0.001) | ||
| G10 | 0.043 ± 0.005 | 0.044 ± 0.006 | 0.046 ± 0.006 | 0.036 ± 0.005 | 0.047 ± 0.006 | 0.031 ± 0.003 | 0.040 ± 0.005 | 0.039 ± 0.005 | 0.038 ± 0.006 | 0.010 (±0.002) | |
| G12 | 0.038 ± 0.006 | 0.039 ± 0.006 | 0.041 ± 0.004 | 0.025 ± 0.005 | 0.038 ± 0.006 | 0.037 ± 0.006 | 0.027 ± 0.005 | 0.034 ± 0.006 | 0.033 ± 0.006 | 0.039 ± 0.006 | 0.001 (±0.001) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultanov, A.; Rola-Łuszczak, M.; Mamanova, S.; Ryło, A.; Osiński, Z.; Saduakassova, M.A.; Bashenova, E.; Kuźmak, J. Molecular Characterization of Bovine Leukemia Virus with the Evidence of a New Genotype Circulating in Cattle from Kazakhstan. Pathogens 2022, 11, 180. https://doi.org/10.3390/pathogens11020180
Sultanov A, Rola-Łuszczak M, Mamanova S, Ryło A, Osiński Z, Saduakassova MA, Bashenova E, Kuźmak J. Molecular Characterization of Bovine Leukemia Virus with the Evidence of a New Genotype Circulating in Cattle from Kazakhstan. Pathogens. 2022; 11(2):180. https://doi.org/10.3390/pathogens11020180
Chicago/Turabian StyleSultanov, Akhmetzhan, Marzena Rola-Łuszczak, Saltanat Mamanova, Anna Ryło, Zbigniew Osiński, Meruyert A. Saduakassova, Elvira Bashenova, and Jacek Kuźmak. 2022. "Molecular Characterization of Bovine Leukemia Virus with the Evidence of a New Genotype Circulating in Cattle from Kazakhstan" Pathogens 11, no. 2: 180. https://doi.org/10.3390/pathogens11020180
APA StyleSultanov, A., Rola-Łuszczak, M., Mamanova, S., Ryło, A., Osiński, Z., Saduakassova, M. A., Bashenova, E., & Kuźmak, J. (2022). Molecular Characterization of Bovine Leukemia Virus with the Evidence of a New Genotype Circulating in Cattle from Kazakhstan. Pathogens, 11(2), 180. https://doi.org/10.3390/pathogens11020180