
Real-Time On-Site Diagnosis of Quarantine Pathogens in Plant Tissues by Nanopore-Based Sequencing

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
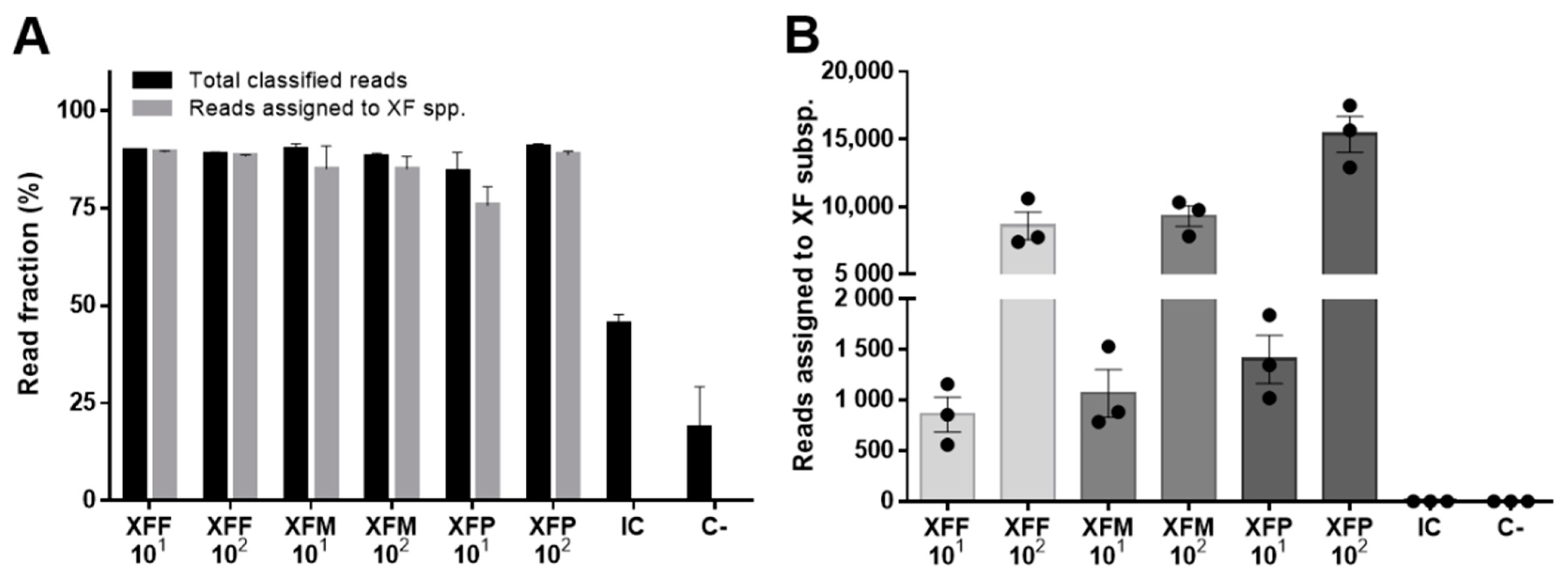
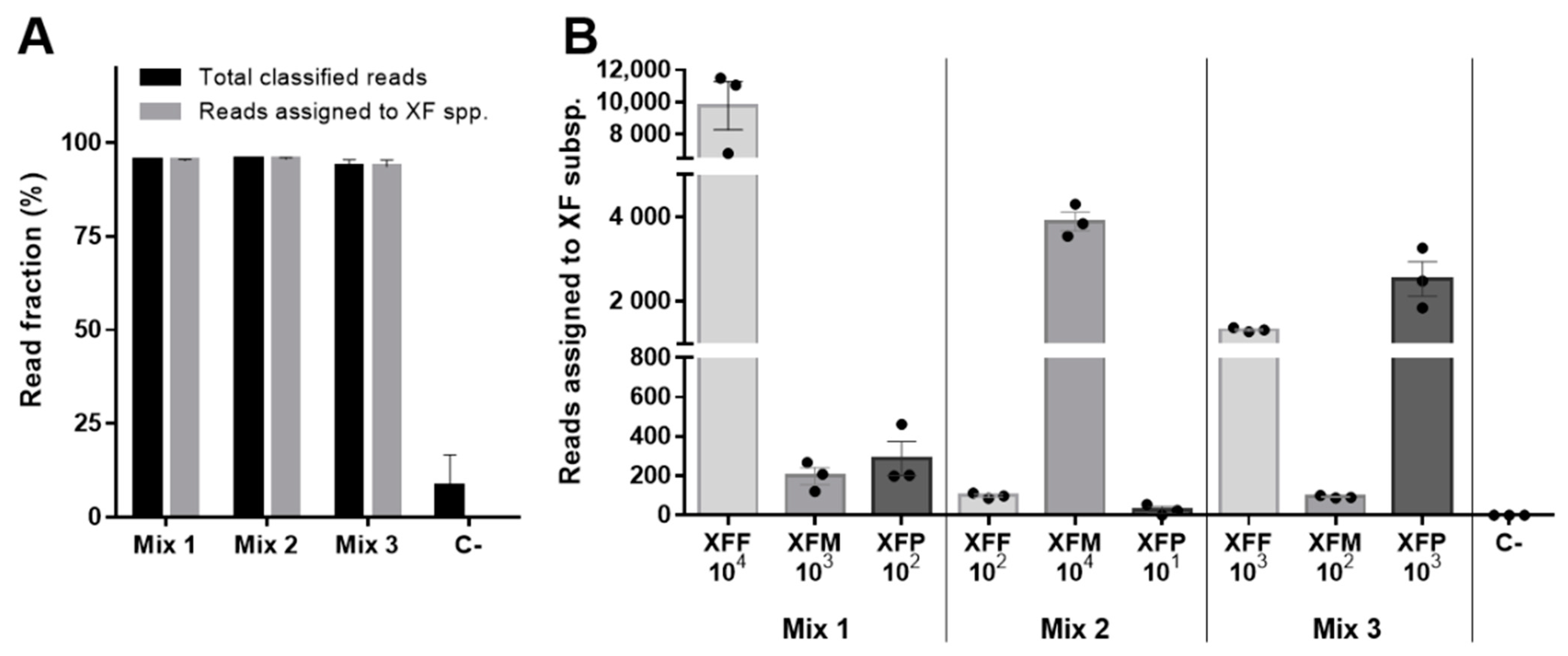
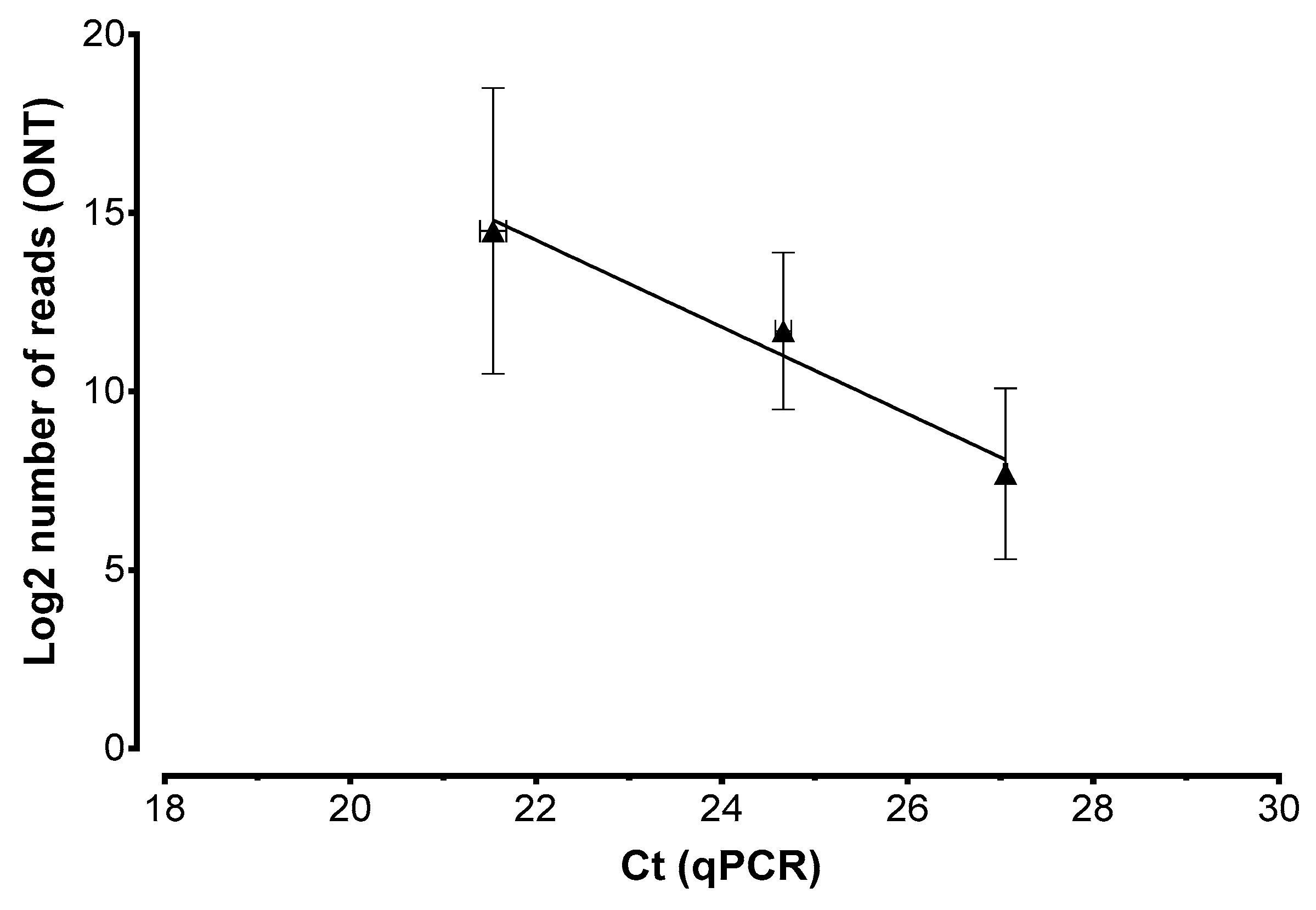
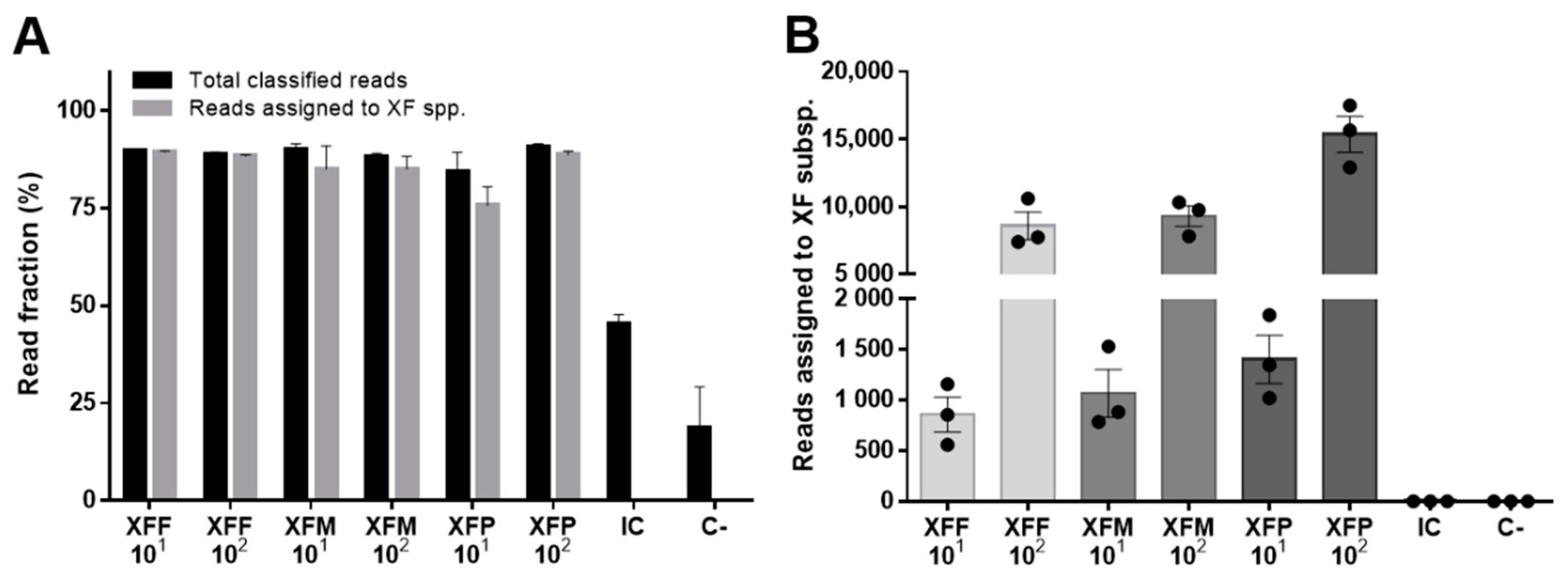
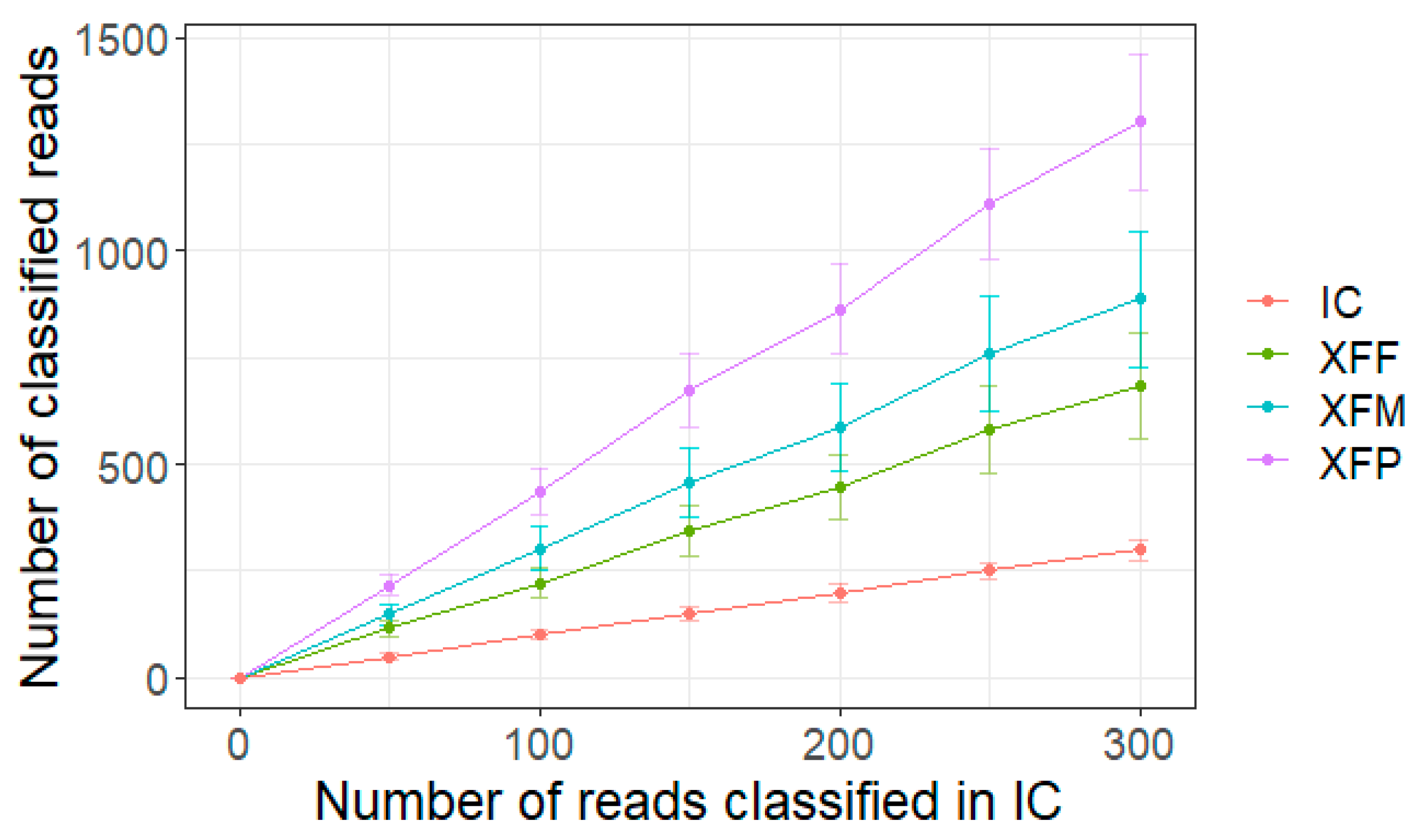
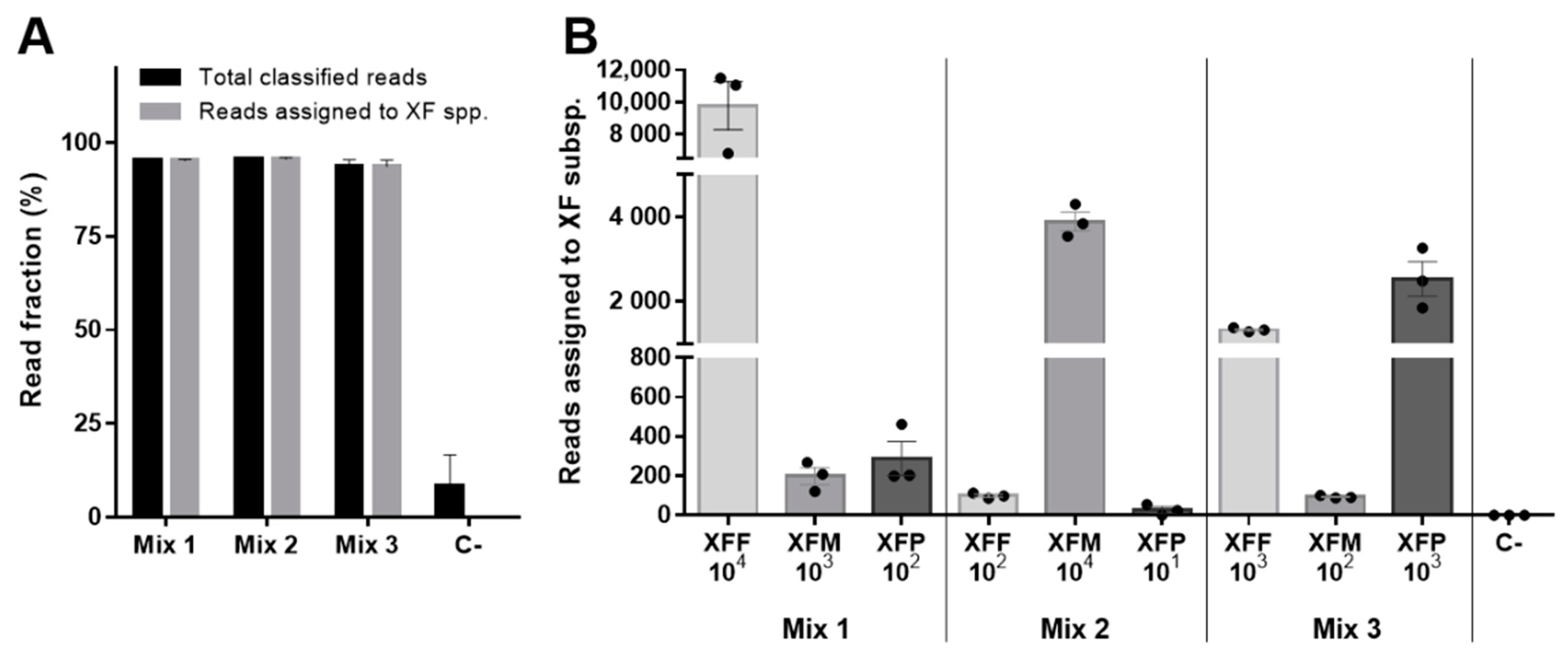
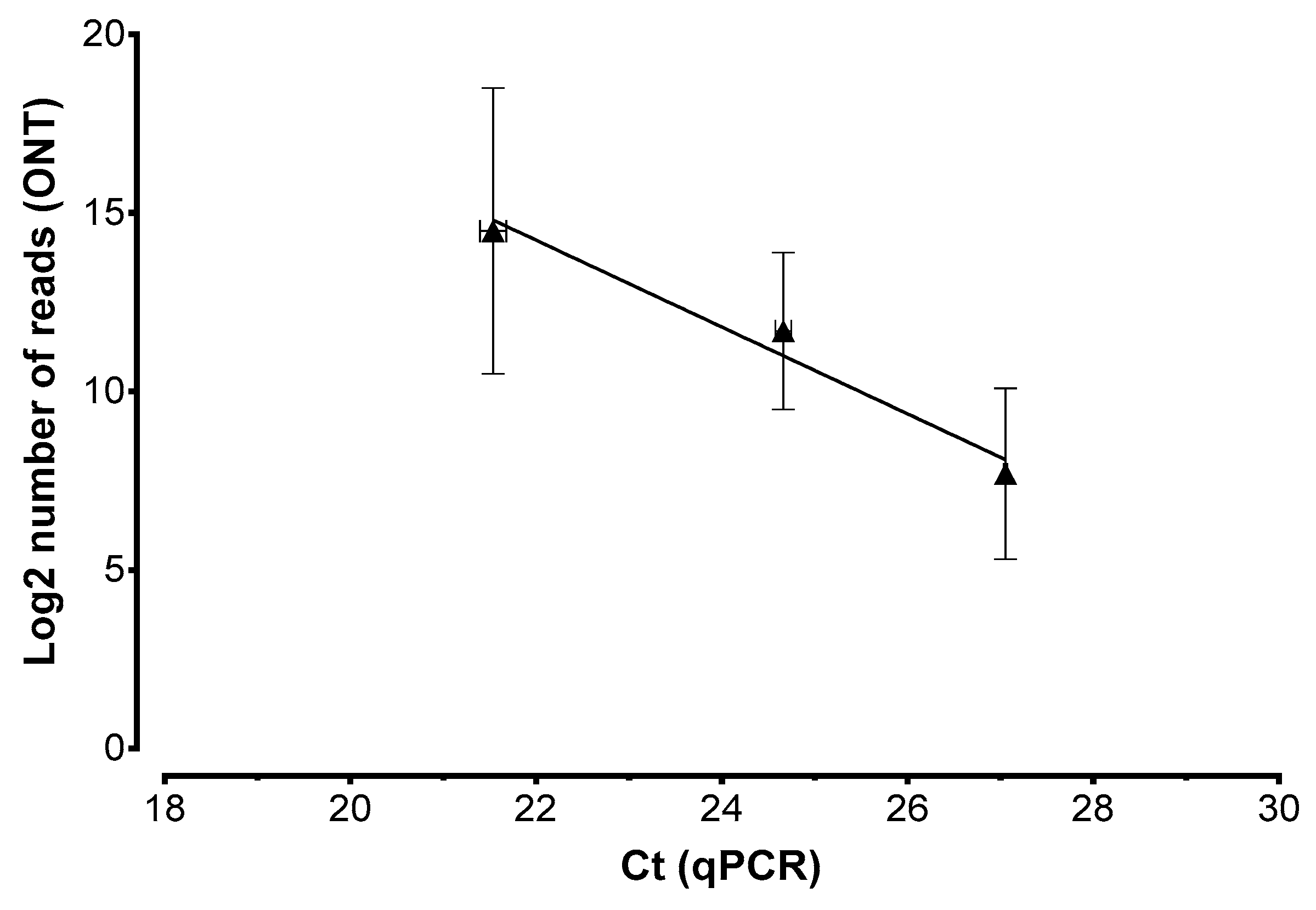
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Amplicon Generation
4.3. ONT Sequencing
4.4. Bioinformatic Analysis
4.5. Real-Time PCR Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aslam, M.N.; Mukhtar, T.; Hussain, M.A.; Raheel, M. Assessment of resistance to bacterial wilt incited by Ralstonia solanacearum in tomato germplasm. J. Plant Dis. Prot. 2017, 124, 585–590. [Google Scholar] [CrossRef]
- Pallás, V.; Sanchez-Navarro, J.A.; James, D. Recent Advances on the Multiplex Molecular Detection of Plant Viruses and Viroids. Front. Microbiol. 2018, 9, 2087. [Google Scholar] [CrossRef] [Green Version]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Torchin, M.E.; Mitchell, C.E. Parasites, pathogens, and invasions by plants and animals. Front. Ecol. Environ. 2004, 2, 183–190. [Google Scholar] [CrossRef]
- Ghelardini, L.; Luchi, N.; Pecori, F.; Pepori, A.L.; Danti, R.; Della Rocca, G.; Capretti, P.; Tsopelas, P.; Santini, A. Ecology of invasive forest pathogens. Biol. Invasions 2017, 19, 3183–3200. [Google Scholar] [CrossRef]
- Schmidhuber, J.; Tubiello, F.N. Global food security under climate change. Proc. Natl. Acad. Sci. USA 2007, 104, 19703–19708. [Google Scholar] [CrossRef] [Green Version]
- La Porta, N.; Capretti, P.; Thomsen, I.M.; Kasanen, R.; Hietala, A.M.; Von Weissenberg, K. Forest pathogens with higher damage potential due to climate change in Europe. Can. J. Plant Pathol. 2008, 30, 177–195. [Google Scholar] [CrossRef]
- Dupas, E.; Legendre, B.; Olivier, V.; Poliakoff, F.; Manceau, C.; Cunty, A. Comparison of real-time PCR and droplet digital PCR for the detection of Xylella fastidiosa in plants. J. Microbiol. Methods 2019, 162, 86–95. [Google Scholar] [CrossRef]
- NAPPPO. Guidelines for the Movement of Propagative Plant Material of Stone Fruit, Pome Fruit, and Grapevine into a NAPPO Member Country; NAPPPO: Ottawa, Canada, 2004. [Google Scholar]
- Baltrus, D.A.; McCann, H.C.; Guttman, D.S. Evolution, genomics and epidemiology of Pseudomonas syringae. Mol. Plant Pathol. 2017, 18, 152–168. [Google Scholar] [CrossRef] [Green Version]
- OEPP/EPPO. EPPO Standard PM 1/2 (30) EPPO A1 and A2 Lists of Pests Recommended for Regulation as Quarantine Pests; EPPO: Klang, Malaysia, 2021. [Google Scholar]
- Candidatus Phytoplasma Solani. 2021. Available online: https://gd.eppo.int/taxon/PHYPSO/datasheet (accessed on 15 February 2021).
- Authority, E.F.S. Update of a database of host plants of Xylella fastidiosa: 20 November 2015. EFSA J. 2016, 14, 4378. [Google Scholar]
- Sicard, A.; Zeilinger, A.R.; Vanhove, M.; Schartel, T.E.; Beal, D.J.; Daugherty, M.P.; Almeida, R.P. Xylella fastidiosa: Insights into an Emerging Plant Pathogen. Annu. Rev. Phytopathol. 2018, 56, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Potnis, N.; Kandel, P.P.; Merfa, M.V.; Retchless, A.C.; Parker, J.K.; Stenger, D.C.; Almeida, R.P.P.; Bergsma-Vlami, M.; Westenberg, M.; Cobine, P.A.; et al. Patterns of inter- and intrasubspecific homologous recombination inform eco-evolutionary dynamics of Xylella fastidiosa. ISME J. 2019, 13, 2319–2333. [Google Scholar] [CrossRef]
- Vanhove, M.; Retchless, A.C.; Sicard, A.; Rieux, A.; Coletta-Filho, H.; De La Fuente, L.; Stenger, D.C.; Almeida, R.P.P. Genomic Diversity and Recombination among Xylella fastidiosa Subspecies. Appl. Environ. Microbiol. 2019, 85, e02972-18. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Purcell, A.H.; Thomson, S.V. Pierce’s Disease of Grapevines: Isolation of the Causal Bacterium. Science 1978, 199, 75–77. [Google Scholar] [CrossRef]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2016, 243, 16–24. [Google Scholar] [CrossRef]
- Piombo, E.; Abdelfattah, A.; Droby, S.; Wisniewski, M.; Spadaro, D.; Schena, L. Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens. Microorganisms 2021, 9, 188. [Google Scholar] [CrossRef]
- Román-Reyna, V.; Dupas, E.; Cesbron, S.; Marchi, G.; Campigli, S.; Hansen, M.A.; Bush, E.; Prarat, M.; Shiplett, K.; Ivey, M.L.L.; et al. Metagenomic Sequencing for Identification of Xylella fastidiosa from Leaf Samples. mSystems 2021, 6, e00591-21. [Google Scholar] [CrossRef]
- Quick, J.; Ashton, P.; Calus, S.T.; Chatt, C.; Gossain, S.; Hawker, J.; Nair, S.; Neal, K.; Nye, K.; Peters, T.; et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 2015, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Quinlan, A.R.; Loman, N.J. A reference bacterial genome dataset generated on the MinION™ portable single-molecule nanopore sequencer. GigaScience 2014, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the Nanopore sequencing platform. Plant Pathol. 2018, 68, 229–238. [Google Scholar] [CrossRef]
- Kilianski, A.; Haas, J.L.; Corriveau, E.J.; Liem, A.T.; Willis, K.L.; Kadavy, D.R.; Rosenzweig, C.N.; Minot, S.S. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. GigaScience 2015, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Keane, J.A. Rapid multi-locus sequence typing direct from uncorrected long reads using Krocus. PeerJ 2018, 6, e5233. [Google Scholar] [CrossRef] [Green Version]
- Faino, L.; Scala, V.; Albanese, A.; Modesti, V.; Grottoli, A.; Pucci, N.; Doddi, A.; L’Aurora, A.; Tatulli, G.; Reverberi, M.; et al. Nanopore sequencing for the detection and identification of Xylella fastidiosa subspecies and sequence types from naturally infected plant material. Plant Pathol. 2021, 70, 1860–1870. [Google Scholar] [CrossRef]
- Krehenwinkel, H.; Kennedy, S.R.; Adams, S.A.; Stephenson, G.T.; Roy, K.; Gillespie, R.G. Multiplex PCR targeting lineage-specific SNP s: A highly efficient and simple approach to block out predator sequences in molecular gut content analysis. Methods Ecol. Evol. 2019, 10, 982–993. [Google Scholar] [CrossRef]
- Maestri, S.; Cosentino, E.; Paterno, M.; Freitag, H.; Garces, J.M.; Marcolungo, L.; Alfano, M.; Njunjić, I.; Schilthuizen, M.; Slik, F.; et al. A Rapid and Accurate MinION-Based Workflow for Tracking Species Biodiversity in the Field. Genes 2019, 10, 468. [Google Scholar] [CrossRef] [Green Version]
- Krehenwinkel, H.; Pomerantz, A.; Prost, S. Genetic Biomonitoring and Biodiversity Assessment Using Portable Sequencing Technologies: Current Uses and Future Directions. Genes 2019, 10, 858. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, A.; Peñafiel, N.; Arteaga, A.; Bustamante, L.; Pichardo, F.; Coloma, A.L.; Barrio-Amorós, C.L.; Salazar-Valenzuela, D.; Prost, S. Real-time DNA barcoding in a rainforest using nanopore sequencing: Opportunities for rapid biodiversity assessments and local capacity building. GigaScience 2018, 7, giy033. [Google Scholar] [CrossRef] [Green Version]
- Watsa, M.; Erkenswick, G.A.; Pomerantz, A.; Prost, S. Portable sequencing as a teaching tool in conservation and biodiversity research. PLoS Biol. 2020, 18, e3000667. [Google Scholar] [CrossRef]
- Faria, N.R.; Quick, J.; Claro, I.; Thézé, J.; De Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; Da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Dupas, E.; Briand, M.; Jacques, M.-A.; Cesbron, S. Novel Tetraplex Quantitative PCR Assays for Simultaneous Detection and Identification of Xylella fastidiosa Subspecies in Plant Tissues. Front. Plant Sci. 2019, 10, 1732. [Google Scholar] [CrossRef] [Green Version]
- Harper, S.J.; Ward, L.I.; Clover, G.R.G. Development of LAMP and Real-Time PCR Methods for the Rapid Detection of Xylella fastidiosa for Quarantine and Field Applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef]
- PM 7/24 (4) Xylella fastidiosa. EPPO Bull. 2019, 49, 175–227. [CrossRef] [Green Version]
- Milián-García, Y.; Young, R.; Madden, M.; Bullas-Appleton, E.; Hanner, R.H. Optimization and validation of a cost-effective protocol for biosurveillance of invasive alien species. Ecol. Evol. 2021, 11, 1999–2014. [Google Scholar] [CrossRef]
- Mang, S.M.; Frisullo, S.; Elshafie, H.S.; Camele, I. Diversity Evaluation of Xylella fastidiosa from Infected Olive Trees in Apulia (Southern Italy). Plant Pathol. J. 2016, 32, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 2020, 39, 442–450. [Google Scholar] [CrossRef]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat. Methods 2021, 18, 165–169. [Google Scholar] [CrossRef]
- Baloğlu, B.; Chen, Z.; Elbrecht, V.; Braukmann, T.; MacDonald, S.; Steinke, D. A workflow for accurate metabarcoding using nanopore MinION sequencing. Methods Ecol. Evol. 2021, 12, 794–804. [Google Scholar] [CrossRef]
- Chen, J.; Groves, R.; Civerolo, E.L.; Viveros, M.; Freeman, M.; Zheng, Y. Two Xylella fastidiosa Genotypes Associated with Almond Leaf Scorch Disease on the Same Location in California. Phytopathology 2005, 95, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Bergsma-Vlami, M.; Van de Bilt, J.L.; Tjou-Tam-Sin, N.N.; Helderman, C.M.; Gorkink-Smits, P.P.; Landman, N.M.; Van Nieuwburg, J.G.; Van Veen, E.J.; Westenberg, M. Assessment of the genetic diversity of Xylella fastidiosa in imported ornamental Coffea arabica plants. Plant Pathol. 2017, 66, 1065–1074. [Google Scholar] [CrossRef]
- Denance, N.; Legendre, B.; Briand, M.; Olivier, V.; De Boisseson, C.; Poliakoff, F.; Jacques, M.A. Several subspecies and sequence types are associated with the emergence of Xylella fastidiosa in natural settings in France. Plant Pathol. 2017, 66, 1054–1064. [Google Scholar] [CrossRef] [Green Version]
- Macho, A.P.; Zumaquero, A.; Ortiz-Martín, I.; Beuzón, C.R. Competitive index in mixed infections: A sensitive and accurate assay for the genetic analysis of Pseudomonas syringae—Plant interactions. Mol. Plant Pathol. 2007, 8, 437–450. [Google Scholar] [CrossRef]
- Vereecke, N.; Bokma, J.; Haesebrouck, F.; Nauwynck, H.; Boyen, F.; Pardon, B.; Theuns, S. High quality genome assemblies of Mycoplasma bovis using a taxon-specific Bonito basecaller for MinION and Flongle long-read nanopore sequencing. BMC Bioinform. 2020, 21, 517. [Google Scholar] [CrossRef]
- Chang, J.J.; Ip, Y.C.; Bauman, A.G.; Huang, D. MinION-in-ARMS: Nanopore Sequencing to Expedite Barcoding of Specimen-Rich Macrofaunal Samples From Autonomous Reef Monitoring Structures. Front. Mar. Sci. 2020, 7, 448. [Google Scholar] [CrossRef]
- Marcolungo, L.; Beltrami, C.; Degli Esposti, C.; Lopatriello, G.; Piubelli, C.; Mori, A.; Pomari, E.; Deiana, M.; Scarso, S.; Bisoffi, Z.; et al. ACoRE: Accurate SARS-CoV-2 genome reconstruction for the characterization of intra-host and inter-host viral diversity in clinical samples and for the evaluation of re-infections. Genomics 2021, 113, 1628–1638. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Tange, O. GNU Parallel—The Command-Line Power Tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chamberlain, S.; Szöcs, E. taxize: Taxonomic search and retrieval in R. F1000Research 2013, 2, 191. [Google Scholar] [CrossRef] [Green Version]
- Francis, M.; Lin, H.; Cabrera-La Rosa, J.; Doddapaneni, H.; Civerolo, E.L. Genome-based PCR Primers for Specific and Sensitive Detection and Quantificationof Xylella fastidiosa. Eur. J. Plant Pathol. 2006, 115, 203. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Host DNA | Experimental Spike | ||
|---|---|---|---|---|
| Host | Concentration | Bacterium | Copies/µL | |
| OXFF_101 | NO | 10 ng/µL | XFF | 101 |
| OXFF_102 | NO | 10 ng/µL | XFF | 102 |
| OXFF_103 | NO | 10 ng/µL | XFF | 103 |
| OXFM_101 | NO | 10 ng/µL | XFM | 101 |
| OXFM_102 | NO | 10 ng/µL | XFM | 102 |
| OXFP_101 | NO | 10 ng/µL | XFP | 101 |
| OXFP_102 | NO | 10 ng/µL | XFP | 102 |
| IC | None | None | SARS-CoV-2, amplicon #96 | 3 ng |
| C- | NO | 10 ng/µL | None | - |
| Mix_1 | NO | 10 ng/µL | XFF, XFM, XFP | 104, 103, 102 |
| Mix_2 | NO | 10 ng/µL | XFF, XFM, XFP | 102, 104, 101 |
| Mix_3 | NO | 10 ng/µL | XFF, XFM, XFP | 103, 102, 103 |
| Sample DNA | XF Spike Copies/µL | Total PASS & Demultiplexed Reads | Total Classified Reads | Reads Assigned to XFF |
|---|---|---|---|---|
| NO + XFF | 103 | 23,094 ± 2043 | 21,752 ± 1859 | 21,703 ± 1855 |
| NO + XFF | 102 | 3287 ± 157 | 3092 ± 169 | 3088 ± 168 |
| NO + XFF | 101 | 205 ± 45 | 187 ± 45 | 187 ± 44 |
| NO (C−) | 0 | 4 ± 1 | 0 ± 0 | 0 ± 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcolungo, L.; Passera, A.; Maestri, S.; Segala, E.; Alfano, M.; Gaffuri, F.; Marturano, G.; Casati, P.; Bianco, P.A.; Delledonne, M. Real-Time On-Site Diagnosis of Quarantine Pathogens in Plant Tissues by Nanopore-Based Sequencing. Pathogens 2022, 11, 199. https://doi.org/10.3390/pathogens11020199
Marcolungo L, Passera A, Maestri S, Segala E, Alfano M, Gaffuri F, Marturano G, Casati P, Bianco PA, Delledonne M. Real-Time On-Site Diagnosis of Quarantine Pathogens in Plant Tissues by Nanopore-Based Sequencing. Pathogens. 2022; 11(2):199. https://doi.org/10.3390/pathogens11020199
Chicago/Turabian StyleMarcolungo, Luca, Alessandro Passera, Simone Maestri, Elena Segala, Massimiliano Alfano, Francesca Gaffuri, Giovanni Marturano, Paola Casati, Piero Attilio Bianco, and Massimo Delledonne. 2022. "Real-Time On-Site Diagnosis of Quarantine Pathogens in Plant Tissues by Nanopore-Based Sequencing" Pathogens 11, no. 2: 199. https://doi.org/10.3390/pathogens11020199
APA StyleMarcolungo, L., Passera, A., Maestri, S., Segala, E., Alfano, M., Gaffuri, F., Marturano, G., Casati, P., Bianco, P. A., & Delledonne, M. (2022). Real-Time On-Site Diagnosis of Quarantine Pathogens in Plant Tissues by Nanopore-Based Sequencing. Pathogens, 11(2), 199. https://doi.org/10.3390/pathogens11020199