
ATM Pathway Is Essential for HPV–Positive Human Cervical Cancer-Derived Cell Lines Viability and Proliferation

 , , and
, , and 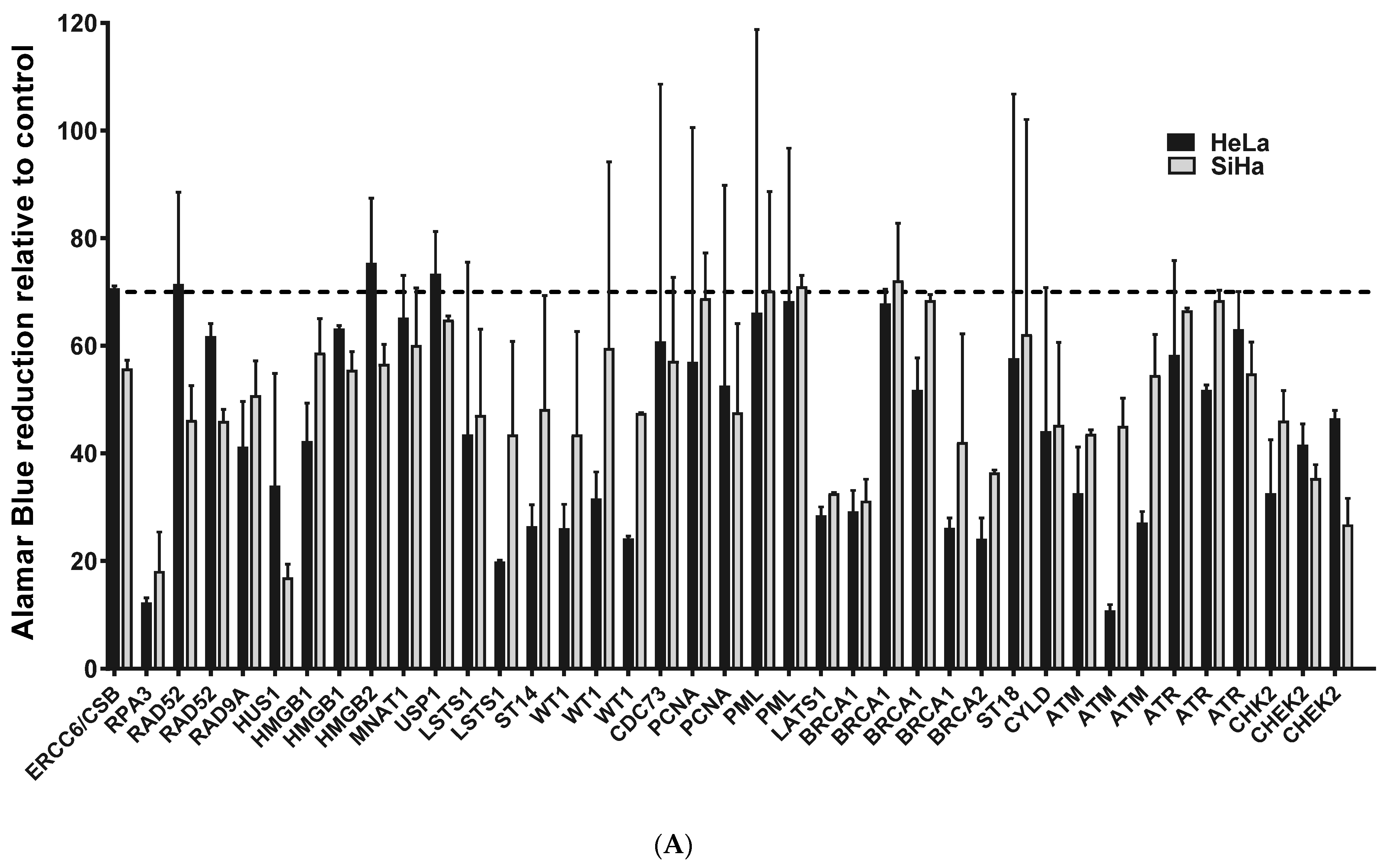{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ATM, BRCA1 and CHEK2 Are Essential for Proliferation/Viability of HPV-Positive Cervical Cancer-Derived Cell Lines
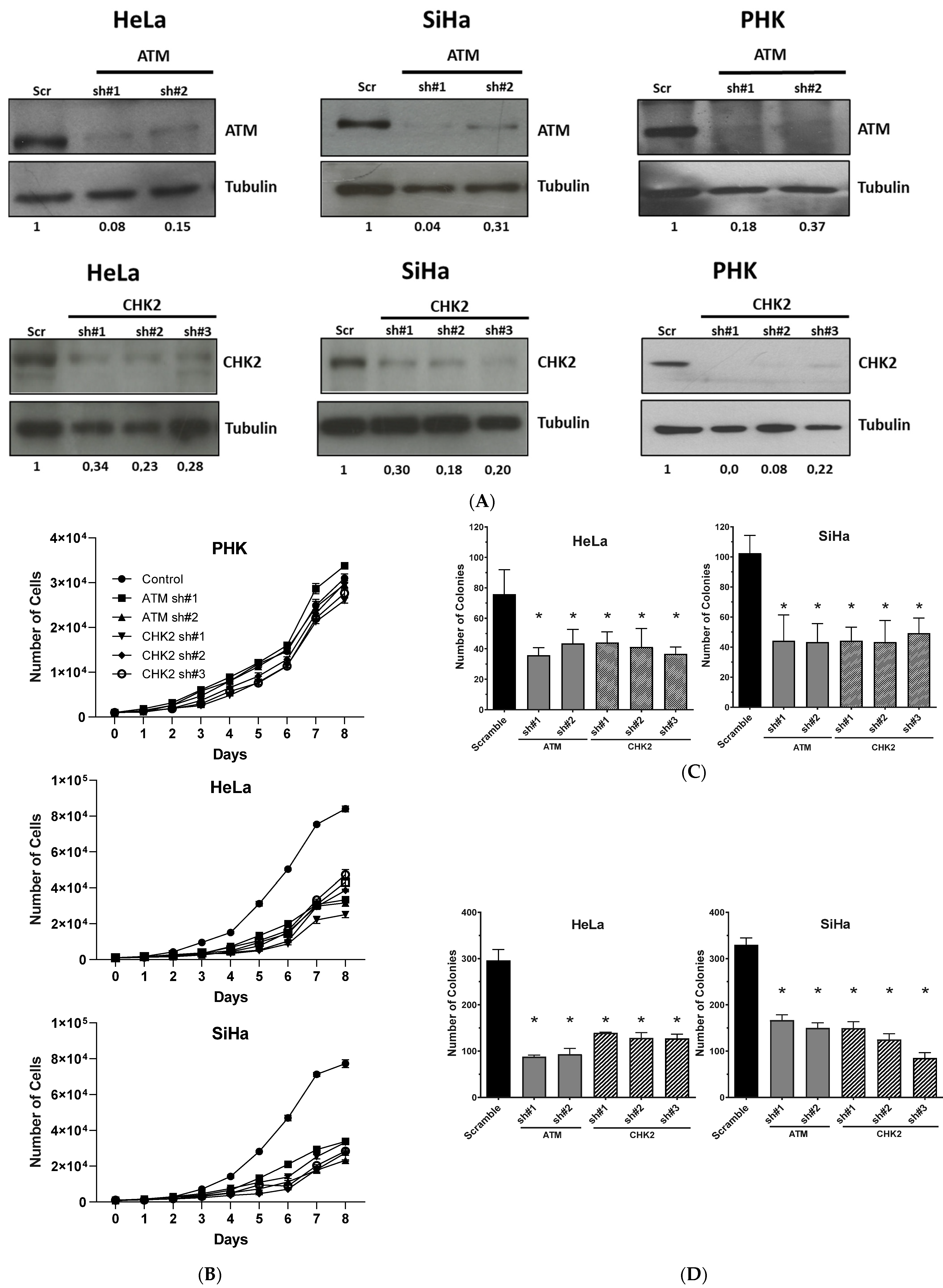
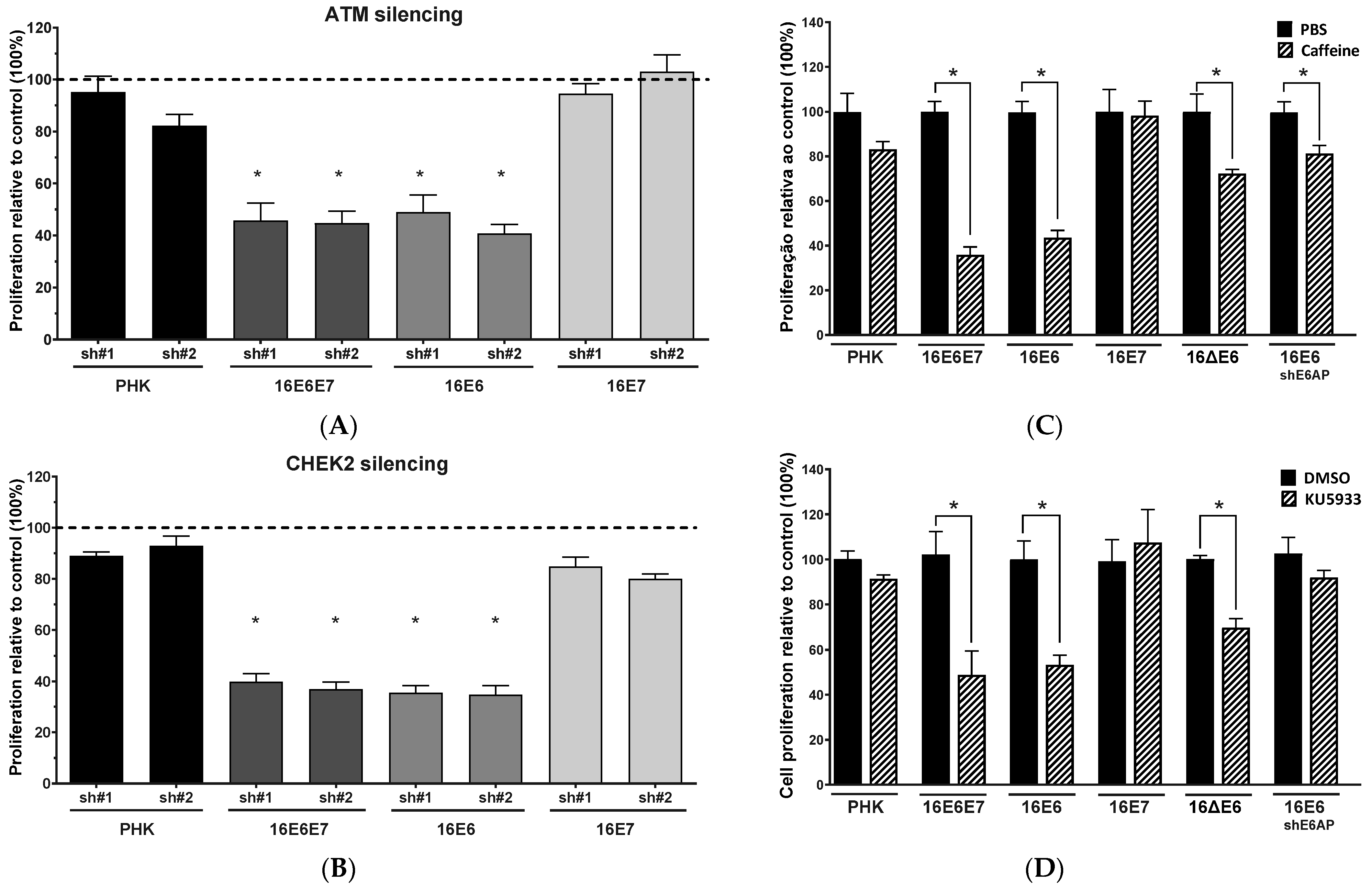
2.2. Gene Silencing or Chemical Inhibition of ATM and CHEK2 Reduces the Proliferation and Clonogenic Capacity of Cervical Cancer-Derived Cells Lines
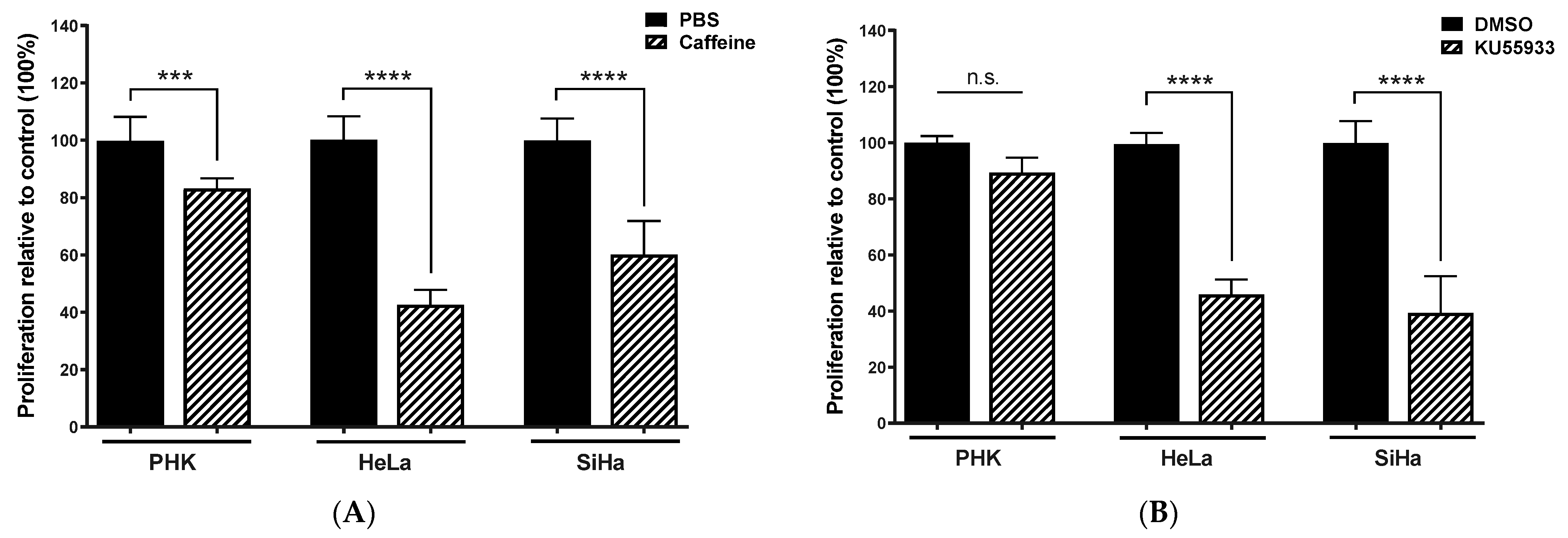
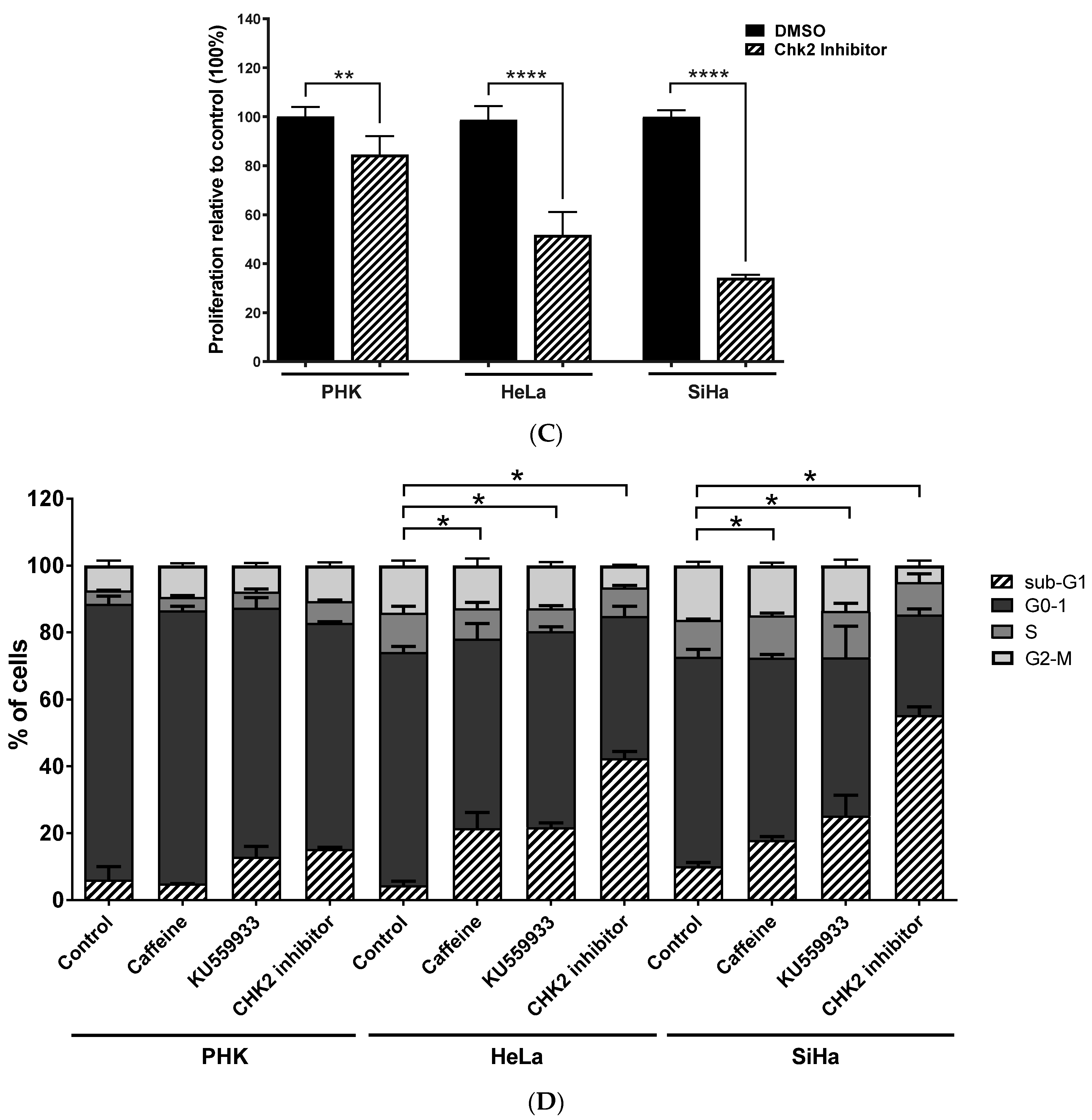
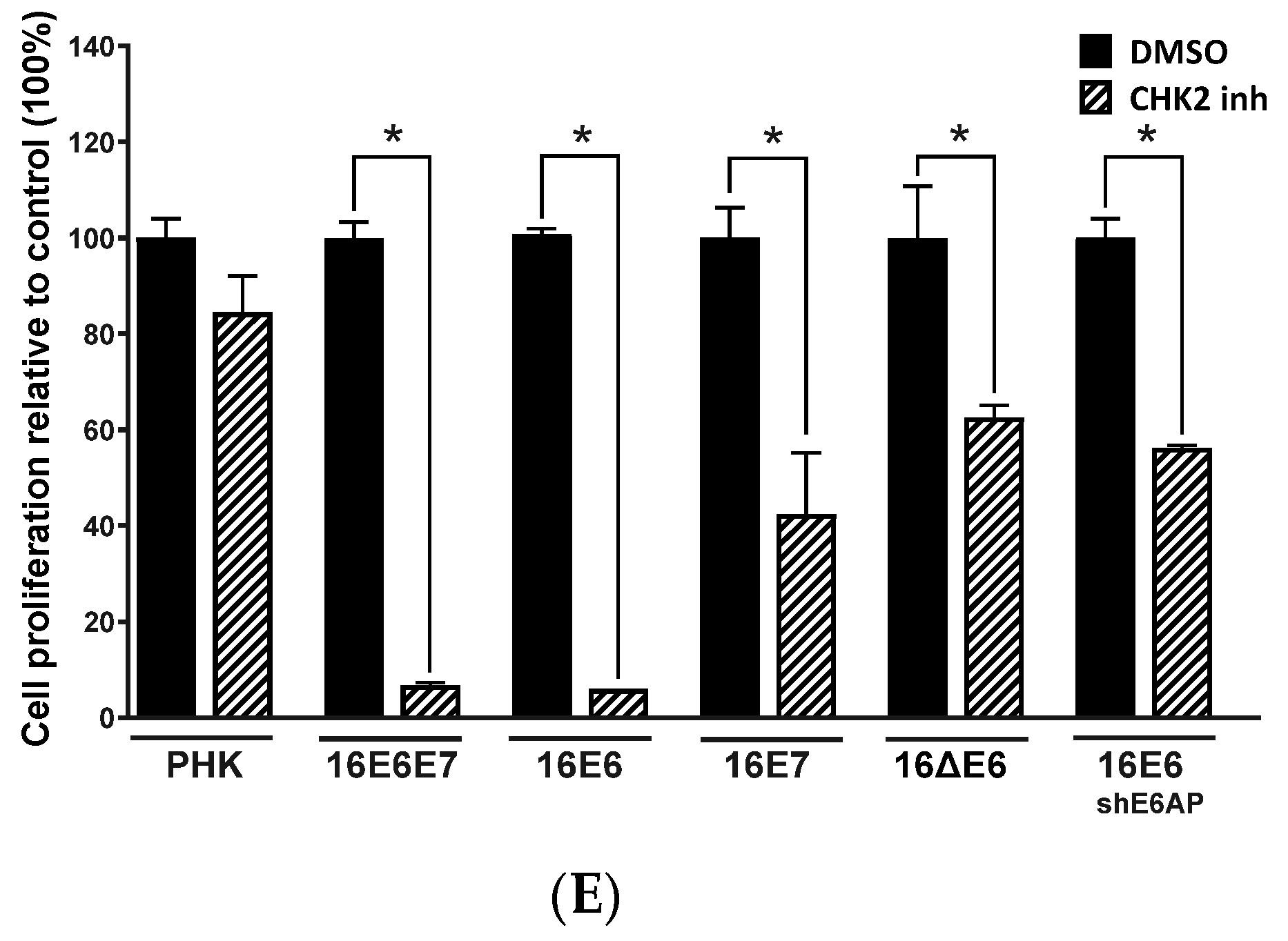
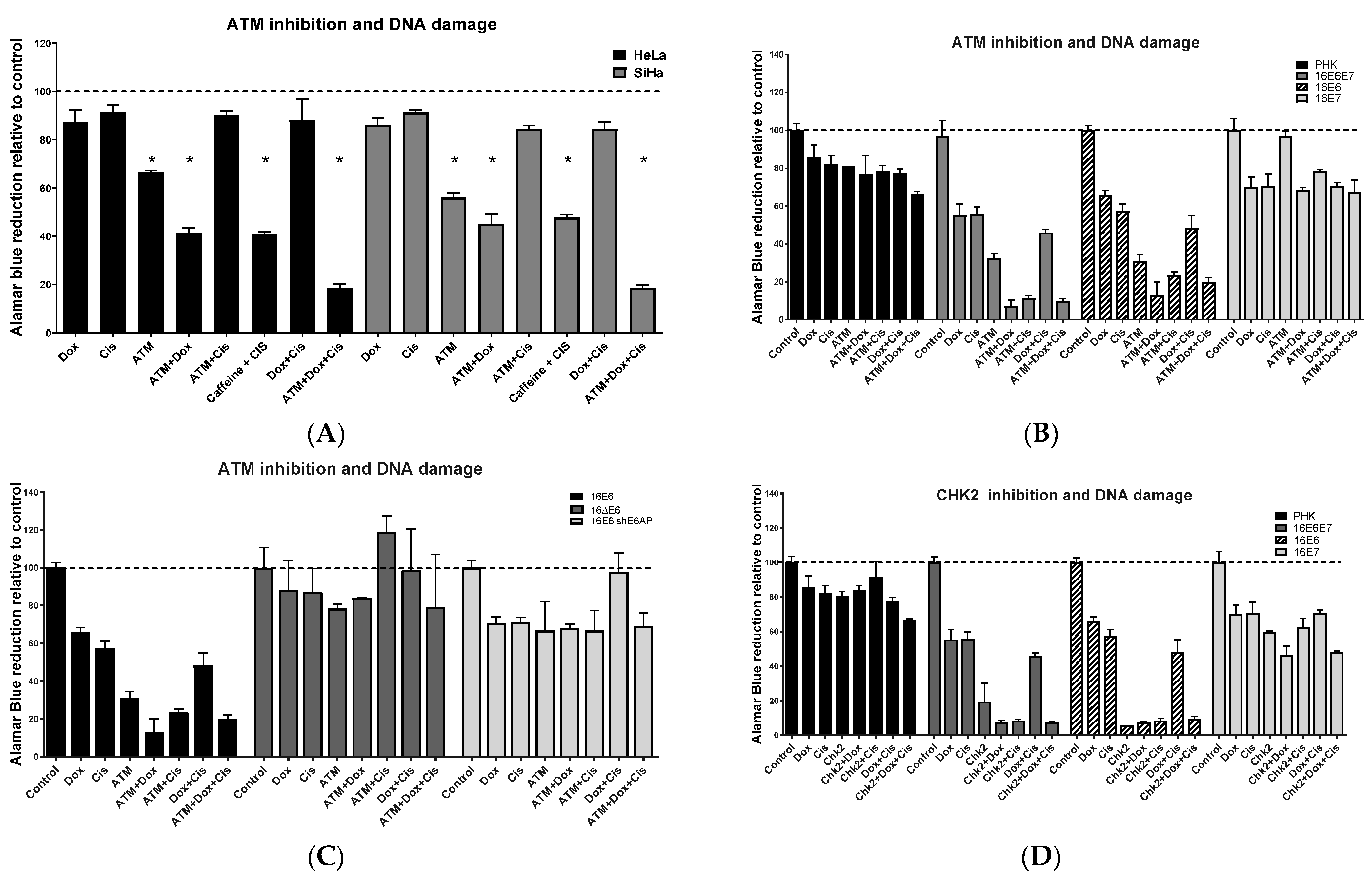
2.3. Chemical Inhibition ATM Reduces Growth Potential and Viability of Cervical Cancer-Derived Cell Lines
2.4. Sensitivity of Cervical Cancer-Derived Cell Lines to ATM and CHK2 Inhibition Depends on the Ability of HPV E6 to Induce p53 Degradation
2.5. ATM/CHEK2 Inhibition Potentiates the Effect of Doxorubicin and Cisplatin in a p53-Dependent Manner
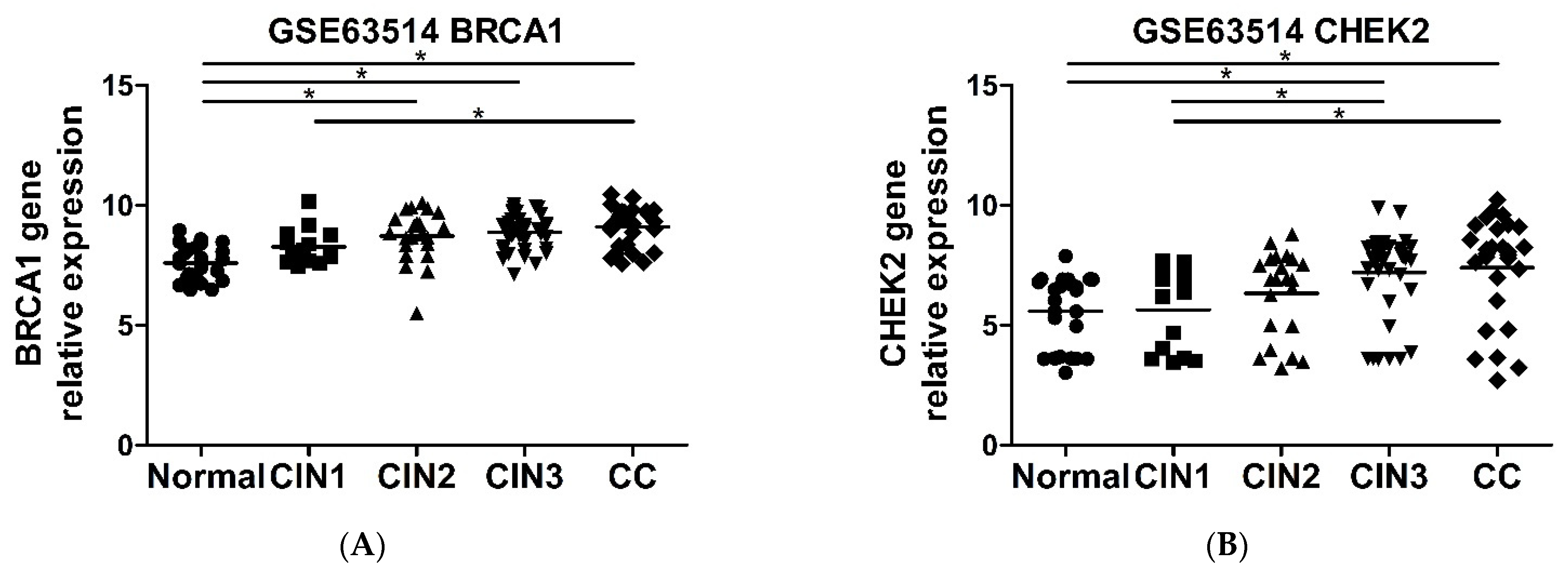
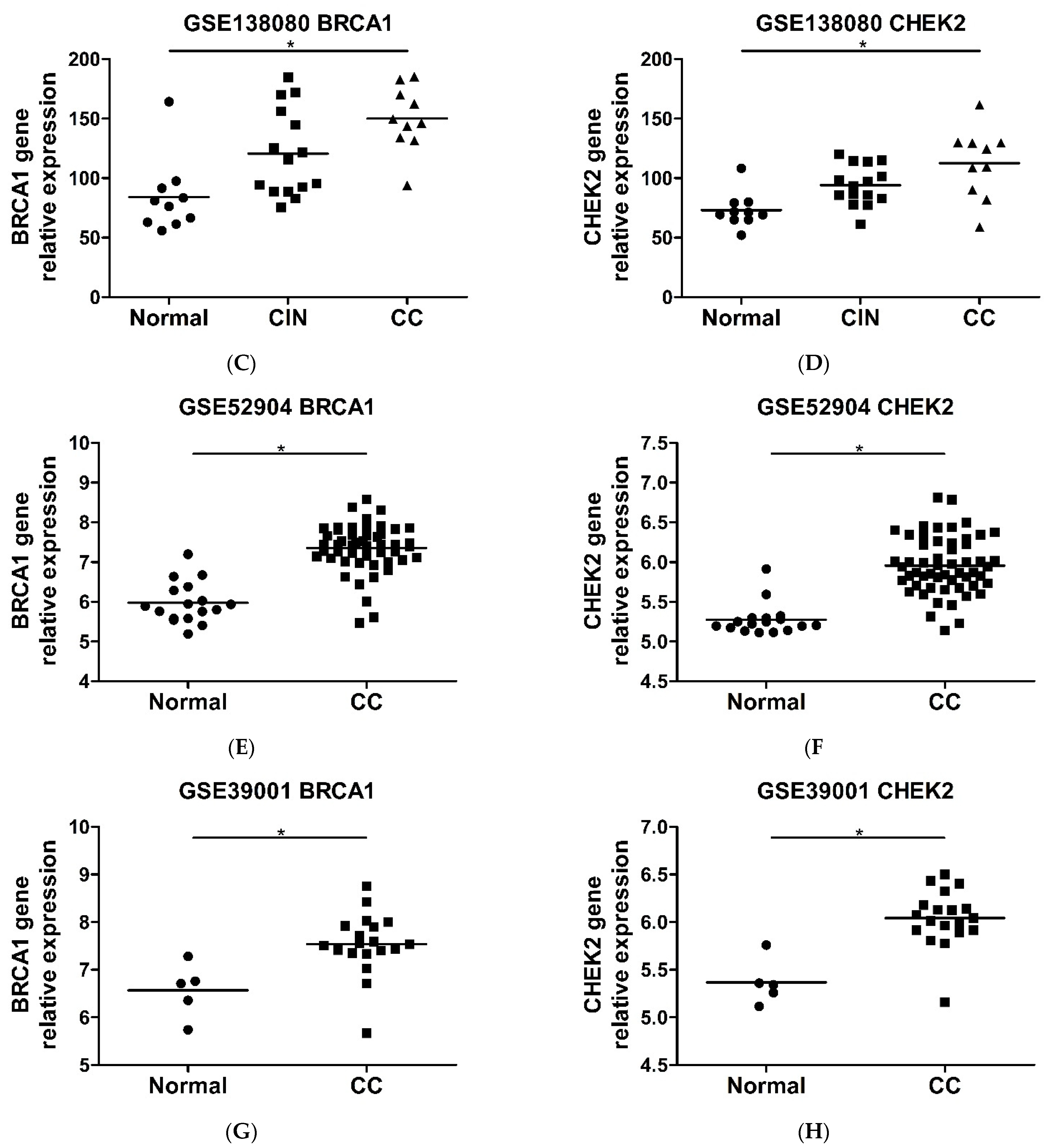
2.6. CHEK2 and BRCA1 Expression Is Higher in Precursor Lesions and Cervical Cancer
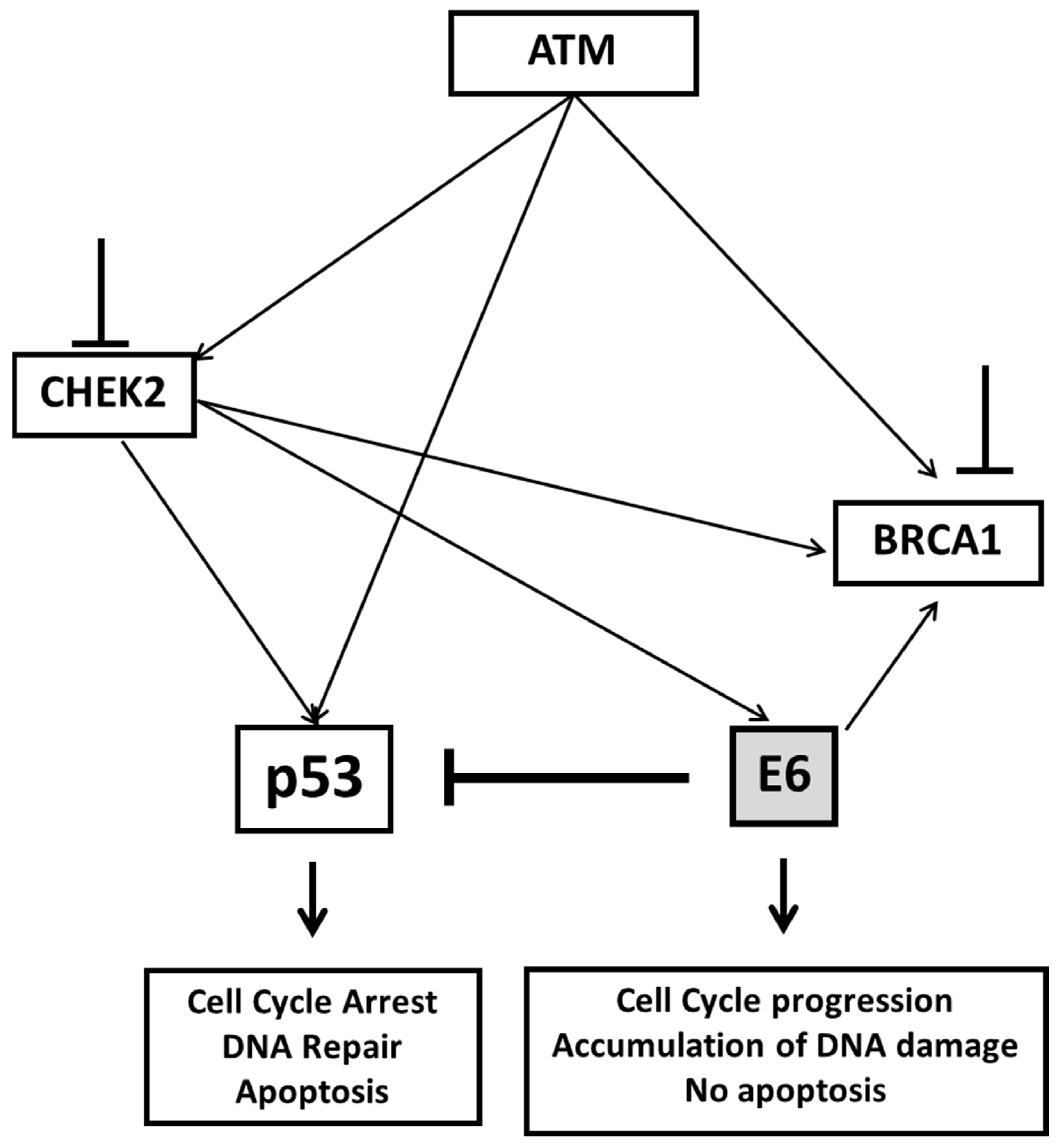
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Retroviruses
4.2. Library Manipulation and DNA Purification
4.3. Lentiviral Virion Production and Quantification
4.4. Infection and Viability Assay
4.5. Cell Proliferation, Clonogenic and Anchorage Independent Growth Assays
4.6. Pharmacological Inhibition ATM and CHK2 and DNA Damage Induction
4.7. Cell Cycle Analysis
4.8. Protein Extraction and Immunoblotting
4.9. In Silico Gene Expression Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Human Papillomaviruses; World Health Organization: Geneva, Switzerland, 2007; Volume 90, pp. 1–636.
- Tota, J.E.; Chevarie-Davis, M.; Richardson, L.A.; DeVries, M.; Franco, E.L. Epidemiology and Burden of HPV Infection and Related Diseases: Implications for Prevention Strategies. Prev. Med. 2011, 53, S12–S21. [Google Scholar] [CrossRef] [PubMed]
- Pett, M.R.; Alazawi, W.O.F.; Roberts, I.; Dowen, S.; Smith, D.I.; Stanley, M.A.; Coleman, N. Acquisition of High-Level Chromosomal Instability Is Associated with Integration of Human Papillomavirus Type 16 in Cervical Keratinocytes. Cancer Res. 2004, 64, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, I.; Algeciras-Schimnich, A.; Song, M.; Sitailo, S.; Policht, F.; Kipp, B.R.; Voss, J.S.; Halling, K.C.; Ruth, A.; King, W.; et al. Chromosomal Biomarkers for Detection of Human Papillomavirus Associated Genomic Instability in Epithelial Cells of Cervical Cytology Specimens. J. Mol. Diagn. 2007, 9, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méhes, G.; Speich, N.; Bollmann, M.; Bollmann, R. Chromosomal Aberrations Accumulate in Polyploid Cells of High-Grade Squamous Intraepithelial Lesions (HSIL). Pathol. Oncol. Res. 2004, 10, 142–148. [Google Scholar] [CrossRef]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Gonzaléz-García, M.C.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Valverde, M.; Rojas, E.; Rodríguez-Sastre, M.A.; García-Cuellar, C.M.; Lizano, M. Human Papillomavirus Types 16 and 18 Early-Expressed Proteins Differentially Modulate the Cellular Redox State and DNA Damage. Int. J. Biol. Sci. 2018, 14, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristol, M.L.; Wang, X.; Smith, N.W.; Son, M.P.; Evans, M.R.; Morgan, I.M. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses 2016, 8, 175. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic Instability of the Host Cell Induced by the Human Papillomavirus Replication Machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of Genomic Instability in Cells Infected with the High-Risk Human Papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- Warburton, A.; Della Fera, A.N.; McBride, A.A. Dangerous Liaisons: Long-Term Replication with an Extrachromosomal HPV Genome. Viruses 2021, 13, 1846. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; Das, D.; Morgan, I.M. Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses 2017, 9, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, A.; Trejo-Cerro, O.; Thomas, M.; Banks, L. Human Papillomavirus E6 and E7: What Remains? Tumour Virus Res. 2021, 11, 200213. [Google Scholar] [CrossRef] [PubMed]
- Scarth, J.A.; Patterson, M.R.; Morgan, E.L.; Macdonald, A. The Human Papillomavirus Oncoproteins: A Review of the Host Pathways Targeted on the Road to Transformation. J. Gen. Virol. 2021, 102, 001540. [Google Scholar] [CrossRef]
- Liu, Y.; Heilman, S.A.; Illanes, D.; Sluder, G.; Chen, J.J. p53-Independent Abrogation of a Postmitotic Checkpoint Contributes to Human Papillomavirus E6-Induced Polyploidy. Cancer Res. 2007, 67, 2603–2610. [Google Scholar] [CrossRef] [Green Version]
- Heilman, S.A.; Nordberg, J.J.; Liu, Y.; Sluder, G.; Chen, J.J. Abrogation of the Postmitotic Checkpoint Contributes to Polyploidization in Human Papillomavirus E7-Expressing Cells. J. Virol. 2009, 83, 2756–2764. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The Human Papillomavirus Type 16 E6 and E7 Oncoproteins Cooperate to Induce Mitotic Defects and Genomic Instability by Uncoupling Centrosome Duplication from the Cell Division Cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Münger, K. The Human Papillomavirus Type 16 E6 and E7 Oncoproteins Independently Induce Numerical and Structural Chromosome Instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar]
- Lembo, D.; Donalisio, M.; Cornaglia, M.; Azzimonti, B.; Demurtas, A.; Landolfo, S. Effect of High-Risk Human Papillomavirus Oncoproteins on p53R2 Gene Expression after DNA Damage. Virus Res. 2006, 122, 189–193. [Google Scholar] [CrossRef]
- Therrien, J.P.; Drouin, R.; Baril, C.; Drobetsky, E.A. Human Cells Compromised for p53 Function Exhibit Defective Global and Transcription-Coupled Nucleotide Excision Repair, Whereas Cells Compromised for pRb Function Are Defective Only in Global Repair. Proc. Natl. Acad. Sci. USA 1999, 96, 15038–15043. [Google Scholar] [CrossRef] [Green Version]
- Kessis, T.D.; Slebos, R.J.; Nelson, W.G.; Kastan, M.B.; Plunkett, B.S.; Han, S.M.; Lorincz, A.T.; Hedrick, L.; Cho, K.R. Human Papillomavirus 16 E6 Expression Disrupts the p53-Mediated Cellular Response to DNA Damage. Proc. Natl. Acad. Sci. USA 1993, 90, 3988–3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morale, M.G.; Da Silva Abjaude, W.; Silva, A.M.; Villa, L.L.; Boccardo, E. HPV-Transformed Cells Exhibit Altered HMGB1-TLR4/MyD88-SARM1 Signaling Axis. Sci. Rep. 2018, 8, 3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.B.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C.M. Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The Human Papilloma Virus-16 E7 Oncoprotein Is Able to Bind to the Retinoblastoma Gene Product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Kurz, E.U.; Douglas, P.; Lees-Miller, S.P. Doxorubicin Activates ATM-Dependent Phosphorylation of Multiple Downstream Targets in Part through the Generation of Reactive Oxygen Species. J. Biol. Chem. 2004, 279, 53272–53281. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lippard, S.J. Cellular Processing of Platinum Anticancer Drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Espinosa, A.M.; Alfaro, A.; Roman-Basaure, E.; Guardado-Estrada, M.; Palma, Í.; Serralde, C.; Medina, I.; Juárez, E.; Bermúdez, M.; Márquez, E.; et al. Mitosis Is a Source of Potential Markers for Screening and Survival and Therapeutic Targets in Cervical Cancer. PLoS ONE 2013, 8, e55975. [Google Scholar] [CrossRef]
- Medina-Martinez, I.; Barrón, V.; Roman-Bassaure, E.; Juárez-Torres, E.; Guardado-Estrada, M.; Espinosa, A.M.; Bermudez, M.; Fernández, F.; Venegas-Vega, C.; Orozco, L.; et al. Impact of Gene Dosage on Gene Expression, Biological Processes and Survival in Cervical Cancer: A Genome-Wide Follow-up Study. PLoS ONE 2014, 9, e55975. [Google Scholar] [CrossRef]
- Den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular Transitions from Papillomavirus Infection to Cervical Precancer and Cancer: Role of Stromal Estrogen Receptor Signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babion, I.; Miok, V.; Jaspers, A.; Huseinovic, A.; Steenbergen, R.D.M.; van Wieringen, W.N.; Wilting, S.M. Identification of Deregulated Pathways, Key Regulators, and Novel miRNA-mRNA Interactions in HPV-Mediated Transformation. Cancers 2020, 12, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogoff, H.A.; Pickering, M.T.; Frame, F.M.; Debatis, M.E.; Sanchez, Y.; Jones, S.; Kowalik, T.F. Apoptosis Associated with Deregulated E2F Activity Is Dependent on E2F1 and Atm/Nbs1/Chk2. Mol. Cell. Biol. 2004, 24, 2968–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J. The Papillomavirus Life Cycle. J. Clin. Virol. 2005, 32 (Suppl. S1), 7. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Liu, Y.; Heilman, S.A.; Chen, J.J. Human Papillomavirus E7 Induces Rereplication in Response to DNA Damage. J. Virol. 2013, 87, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.; Grueneberg, D.A.; Hellner, K.; Sawyer, J.; Grace, M.; Li, W.; Harlow, E.; Munger, K. Kinase Requirements in Human Cells: V. Synthetic Lethal Interactions between p53 and the Protein Kinases SGK2 and PAK3. Proc. Natl. Acad. Sci. USA 2010, 107, 12463–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G. Synthetic Lethality: A Framework for the Development of Wiser Cancer Therapeutics. Genome Med. 2009, 1, 99. [Google Scholar] [CrossRef] [Green Version]
- Christian Reinhardt, H.; Jiang, H.; Hemann, M.T.; Yaffe, M.B. Exploiting Synthetic Lethal Interactions for Targeted Cancer Therapy. Cell Cycle 2009, 8, 3112–3119. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A Genome-Wide RNAi Screen Identifies Multiple Synthetic Lethal Interactions with the Ras Oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Kwok, M.; Agathanggelou, A.; Davies, N.; Stankovic, T. Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives. Cancers 2021, 13, 4681. [Google Scholar] [CrossRef]
- Ashley, A.K.; Kemp, C.J. DNA-PK, ATM, and ATR: PIKKing on p53. Cell Cycle 2018, 17, 275–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Laimins, L.A. The JAK-STAT Transcriptional Regulator, STAT-5, Activates the ATM DNA Damage Pathway to Induce HPV 31 Genome Amplification upon Epithelial Differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusho, E.; Laimins, L. Human Papillomaviruses Target the DNA Damage Repair and Innate Immune Response Pathways to Allow for Persistent Infection. Viruses 2021, 13, 1390. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio 2018, 9, e00064-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fan, S.; Meng, Q.; Ma, Y.; Katiyar, P.; Schlegel, R.; Rosen, E.M. BRCA1 Interaction with Human Papillomavirus Oncoproteins. J. Biol. Chem. 2005, 280, 33165–33177. [Google Scholar] [CrossRef] [Green Version]
- Thatte, J.; Massimi, P.; Thomas, M.; Boon, S.S.; Banks, L. The Human Papillomavirus E6 PDZ Binding Motif Links DNA Damage Response Signaling to E6 Inhibition of p53 Transcriptional Activity. J. Virol. 2018, 92, e00465-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prati, B.; da Silva Abjaude, W.; Termini, L.; Morale, M.; Herbster, S.; Longatto-Filho, A.; Nunes, R.A.L.; Córdoba Camacho, L.C.; Rabelo-Santos, S.H.; Zeferino, L.C.; et al. Three Prime Repair Exonuclease 1 (TREX1) Expression Correlates with Cervical Cancer Cells Growth in Vitro and Disease Progression in Vivo. Sci. Rep. 2019, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Waterman, D.P.; Haber, J.E.; Smolka, M.B. Checkpoint Responses to DNA Double-Strand Breaks. Annu. Rev. Biochem. 2020, 89, 103–133. [Google Scholar] [CrossRef] [Green Version]
- Boccardo, E.; Manzini Baldi, C.V.; Carvalho, A.F.; Rabachini, T.; Torres, C.; Barreta, L.A.; Brentani, H.; Villa, L.L. Expression of Human Papillomavirus Type 16 E7 Oncoprotein Alters Keratinocytes Expression Profile in Response to Tumor Necrosis Factor-α. Carcinogenesis 2010, 31, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Helt, A.-M.; Galloway, D.A. Destabilization of the Retinoblastoma Tumor Suppressor by Human Papillomavirus Type 16 E7 Is Not Sufficient to Overcome Cell Cycle Arrest in Human Keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef] [Green Version]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase Activation by the E6 Gene Product of Human Papillomavirus Type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abjaude, W.; Prati, B.; Munford, V.; Montenegro, A.; Lino, V.; Herbster, S.; Rabachini, T.; Termini, L.; Menck, C.F.M.; Boccardo, E. ATM Pathway Is Essential for HPV–Positive Human Cervical Cancer-Derived Cell Lines Viability and Proliferation. Pathogens 2022, 11, 637. https://doi.org/10.3390/pathogens11060637
Abjaude W, Prati B, Munford V, Montenegro A, Lino V, Herbster S, Rabachini T, Termini L, Menck CFM, Boccardo E. ATM Pathway Is Essential for HPV–Positive Human Cervical Cancer-Derived Cell Lines Viability and Proliferation. Pathogens. 2022; 11(6):637. https://doi.org/10.3390/pathogens11060637
Chicago/Turabian StyleAbjaude, Walason, Bruna Prati, Veridiana Munford, Aline Montenegro, Vanesca Lino, Suellen Herbster, Tatiana Rabachini, Lara Termini, Carlos Frederico Martins Menck, and Enrique Boccardo. 2022. "ATM Pathway Is Essential for HPV–Positive Human Cervical Cancer-Derived Cell Lines Viability and Proliferation" Pathogens 11, no. 6: 637. https://doi.org/10.3390/pathogens11060637
APA StyleAbjaude, W., Prati, B., Munford, V., Montenegro, A., Lino, V., Herbster, S., Rabachini, T., Termini, L., Menck, C. F. M., & Boccardo, E. (2022). ATM Pathway Is Essential for HPV–Positive Human Cervical Cancer-Derived Cell Lines Viability and Proliferation. Pathogens, 11(6), 637. https://doi.org/10.3390/pathogens11060637