
Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study

 ,
,
Abstract
:1. Introduction
2. Results
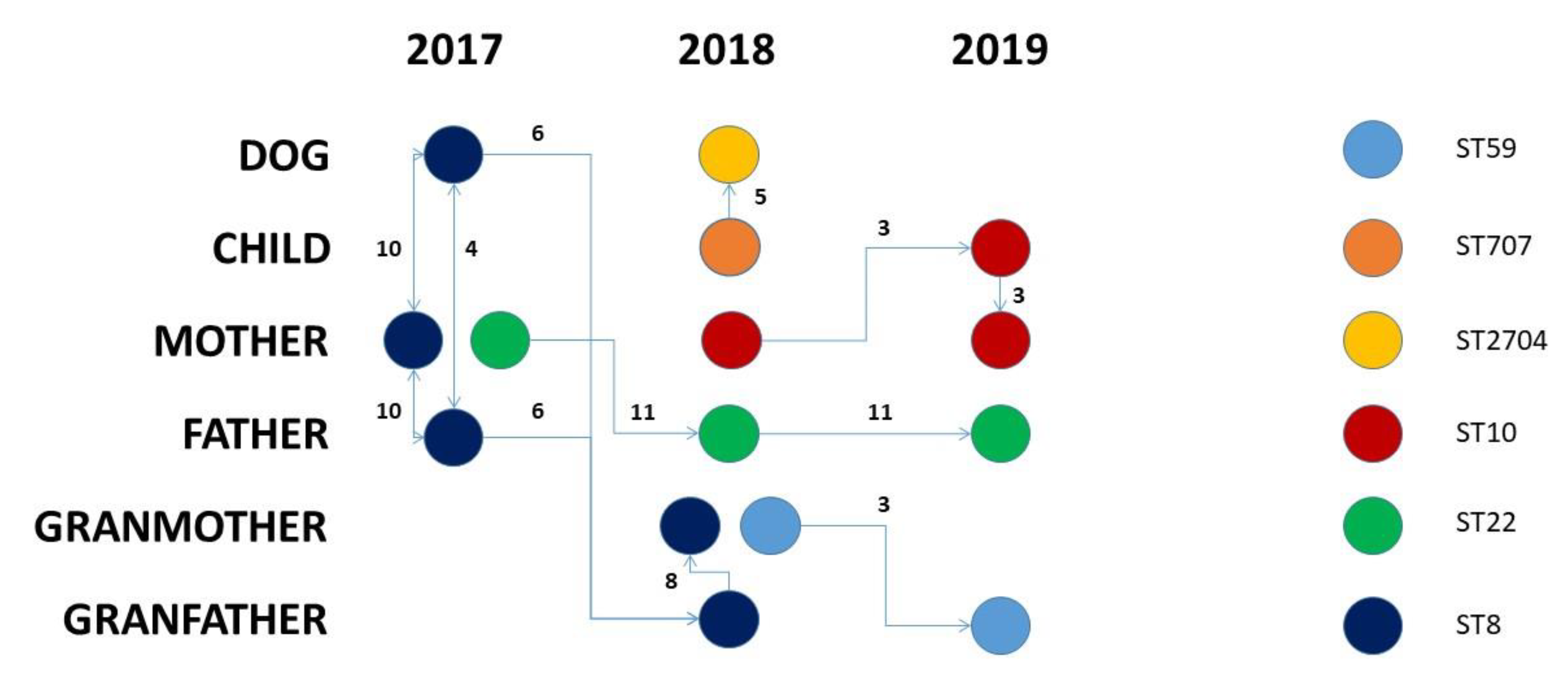
2.1. Clinical Context
2.2. Dataset Description
2.3. Genomic Analysis
2.4. Antibiotic Resistance Profiling
2.5. Prophages Analysis
2.6. Plasmids Content
2.7. IEC Genes
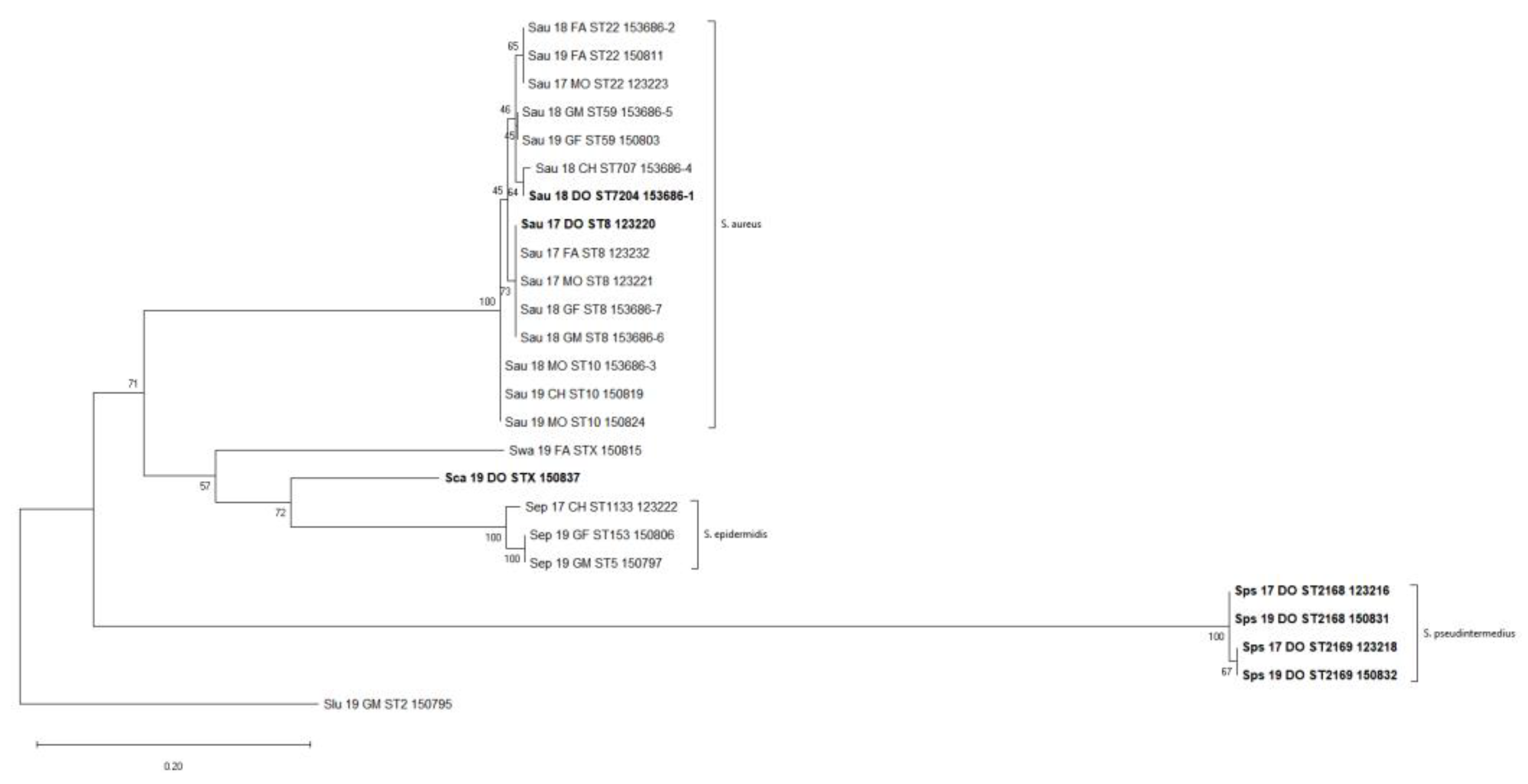
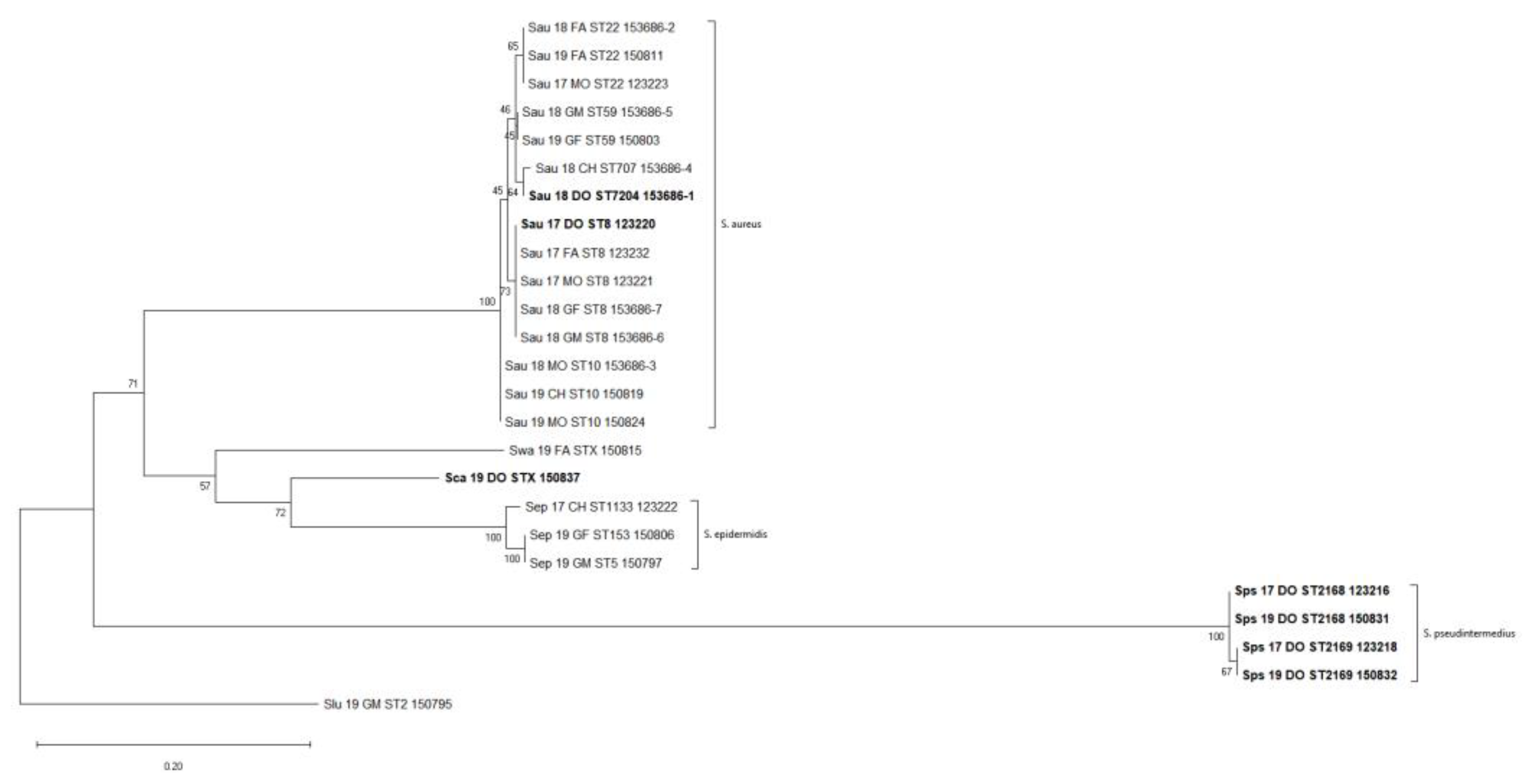
2.8. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Microbiological Investigations
4.3. DNA Extraction and Whole-Genome Sequencing
4.4. Genome Assembly and Annotation
4.5. Accession Number(s)
4.6. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, J.Y.; Seo, K.S. Staphylococcus aureus. In Food Microbiology: Fundamentals and Frontiers; American Society for Microbiology: Washington, DC, USA, 2022; pp. 555–584. [Google Scholar] [CrossRef]
- Hidron, A.I.; Low, C.E.; Honig, E.G.; Blumberg, H.M. Emergence of community-acquired meticillin-resistant Staphylococcus aureus strain USA300 as a cause of necrotising community-onset pneumonia. Lancet Infect. Dis. 2009, 9, 384–392. [Google Scholar] [CrossRef]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4, 18033. [Google Scholar] [CrossRef] [PubMed]
- Petinaki, E.; Spiliopoulou, I. Methicillin-resistant Staphylococcus aureus colonization and infection risks from companion animals: Current perspectives. Vet. Med. Res. Rep. 2015, 6, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Bierowiec, K.; Płoneczka-Janeczko, K.; Rypuła, K. Is the Colonisation of Staphylococcus aureus in Pets Associated with Their Close Contact with Owners? PLoS ONE 2016, 11, e0156052. [Google Scholar] [CrossRef]
- Balasubramanian, D.; Harper, L.; Shopsin, B.; Torres, V.J. Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis. 2017, 75, ftx005. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Cerquetella, M.; Attili, A.R. Amphixenosic Aspects of Staphylococcus aureus Infection in Man and Animals. Curr. Top. Microbiol. Immunol. 2017, 409, 297–323. [Google Scholar] [CrossRef]
- Matuszewska, M.; Murray, G.G.R.; Harrison, E.M.; Holmes, M.A.; Weinert, L.A. The Evolutionary Genomics of Host Specificity in Staphylococcus aureus. Trends Microbiol. 2020, 28, 465–477. [Google Scholar] [CrossRef]
- Yu, F.; Cienfuegos-Gallet, A.V.; Cunningham, M.H.; Jin, Y.; Wang, B.; Kreiswirth, B.N.; Chen, L. Molecular Evolution and Adaptation of Livestock-Associated Methicillin-Resistant Staphylococcus aureus (LA-MRSA) Sequence Type 9. mSystems 2021, 6, e0049221. [Google Scholar] [CrossRef]
- Ahmadrajabi, R.; Layegh-Khavidaki, S.; Kalantar-Neyestanaki, D.; Fasihi, Y. Molecular analysis of immune evasion cluster (IEC) genes and intercellular adhesion gene cluster (ICA) among methicillin-resistant and methicillin-sensitive isolates of Staphylococcus aureus. J. Prev. Med. Hyg. 2017, 58, E308–E314. [Google Scholar] [CrossRef]
- Uhlemann, A.-C.; Dordel, J.; Knox, J.R.; Raven, K.E.; Parkhill, J.; Holden, M.T.G.; Peacock, S.J.; Lowy, F.D. Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York Community. Proc. Natl. Acad. Sci. USA 2014, 111, 6738–6743. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.R. Evolution of Staphylococcus aureus during human colonization and infection. Infect. Genet. Evol. 2014, 21, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Maali, Y.; Badiou, C.; Martins-Simões, P.; Hodille, E.; Bes, M.; Vandenesch, F.; Lina, G.; Diot, A.; Laurent, F.; Trouillet-Assant, S. Understanding the virulence of Staphylococcus pseudintermedius: A major role of pore-forming toxins. Front. Cell. Infect. Microbiol. 2018, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Strauß, L.; Stegger, M.; Akpaka, P.E.; Alabi, A.; Breurec, S.; Coombs, G.; Egyir, B.; Larsen, A.R.; Laurent, F.; Monecke, S.; et al. Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc. Natl. Acad. Sci. USA 2017, 114, E10596–E10604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monecke, S.; Coombs, G.; Shore, A.C.; Coleman, D.C.; Akpaka, P.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A Field Guide to Pandemic, Epidemic and Sporadic Clones of Methicillin-Resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- McClure, J.A.; Lakhundi, S.; Niazy, A.; Dong, G.; Obasuyi, O.; Gordon, P.; Chen, S.; Conly, J.M.; Zhang, K. Staphylococcus aureus ST59: Concurrent but Separate Evolution of North American and East Asian Lineages. Front. Microbiol. 2021, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Higuchi, W.; Otsuka, T.; Baranovich, T.; Enany, S.; Saito, K.; Isobe, H.; Dohmae, S.; Ozaki, K.; Takano, M.; et al. Novel Characteristics of Community-Acquired Methicillin-Resistant Staphylococcus aureus Strains Belonging to Multilocus Sequence Type 59 in Taiwan. Antimicrob. Agents Chemother. 2008, 52, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Rooijakkers, S.H.M.; van Kessel, K.P.M.; van Strijp, J.A.G. Staphylococcal innate immune evasion. Trends Microbiol. 2005, 13, 596–601. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Flowers, L.; Grice, E.A. The Skin Microbiota: Balancing Risk and Reward. Cell Host Microbe 2020, 28, 190–200. [Google Scholar] [CrossRef]
- Bloemendaal, A.L.A.; Brouwer, E.C.; Fluit, A.C. Methicillin Resistance Transfer from Staphylocccus epidermidis to Methicillin-Susceptible Staphylococcus aureus in a Patient during Antibiotic Therapy. PLoS ONE 2010, 5, e11841. [Google Scholar] [CrossRef]
- Louie, L.; Goodfellow, J.; Mathieu, P.; Glatt, A.; Louie, M.; Simor, A.E. Rapid Detection of Methicillin-Resistant Staphylococci from Blood Culture Bottles by Using a Multiplex PCR Assay. J. Clin. Microbiol. 2002, 40, 2786–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling Genomes and Mini-metagenomes from Highly Chimeric Reads. Lect. Notes Comput. Sci. 2013, 7821, 158–170. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid Whole-Genome Sequencing for Detection and Characterization of Microorganisms Directly from Clinical Samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Gardner, S.N.; Slezak, T.; Hall, B.G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 2015, 31, 2877–2878. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Sample | Organism | Collection Date | Family Member, District | ST |
|---|---|---|---|---|
| Sau_17_FA_ST8_123232 | S. aureus | 2017 | father, skin | ST8 |
| Sau_17_MO_ST8_123221 | S. aureus | 2017 | mother, mouth | ST8 |
| Sau_17_DO_ST8_123220 | S. aureus | 2017 | dog, armpit | ST8 |
| Sau_18_GF_ST8_153686-7 | S. aureus | 2018 | grandfather, nose | ST8 |
| Sau_18_GM_ST8_153686-6 | S. aureus | 2018 | grandmother, armpit | ST8 |
| Sau_18_MO_ST10_153686-3 | S. aureus | 2018 | mother, nose | ST10 |
| Sau_19_MO_ST10_150824 | S. aureus | 2019 | mother, nose | ST10 |
| Sau_19_CH_ST10_150819 | S. aureus | 2019 | child, nose | ST10 |
| Sau_17_MO_ST22_123223 | S. aureus | 2017 | mother, nose | ST22 |
| Sau_18_FA_ST22_153686-2 | S. aureus | 2018 | father, nose | ST22 |
| Sau_19_FA_ST22_150811 | S. aureus | 2019 | father, nose | ST22 |
| Sau_18_GM_ST59_153686-5 | S. aureus | 2018 | grandmother, nose | ST59 |
| Sau_19_GF_ST59_150803 | S. aureus | 2019 | grandfather, nose | ST59 |
| Sau_18_CH_ST707_153686-4 | S. aureus | 2018 | child, nose | ST707 |
| Sau_18_DO_ST7204_153686-1 | S. aureus | 2018 | dog, mouth | ST7204 |
| Sca_19_DO_STX_150837 | S. capitis | 2019 | dog, armpit | ND |
| Sep_19_GF_ST153_150806 | S. epidermidis | 2019 | grandfather, nose | ST153 |
| Sep_17_CH_ST1133_123222 | S. epidermidis | 2017 | child, mouth | ST1133 |
| Sep_19_GM_ST5_150797 | S. epidermidis | 2019 | grandmother, nose | ST5 |
| Slu_19_GM_ST2_150795 | S. lugdunensis | 2019 | grandmother, nose | ST2 |
| Sps_17_DO_ST2169_123218 | S. pseudintermedius | 2017 | dog, mouth | ST2169 |
| Sps_19_DO_ST2169_150832 | S. pseudintermedius | 2019 | dog, foreskin | ST2169 |
| Sps_17_DO_ST2168_123216 | S. pseudintermedius | 2017 | dog, mouth | ST2168 |
| Sps_19_DO_ST2168_150831 | S. pseudintermedius | 2019 | dog, mouth | ST2168 |
| Swa_19_FA_STX_150815 | S. warneri | 2019 | father, nose | ND |
| Sample | blaZ | ant(6)-Ia | aph(3)-III | aadD | aac(6)-aph(2) | cat(pC221) | fosB | fusB | mecA | mph(C) | msr(A) | erm(C) | erm(B) | vga(A) | vga(A)LC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sau_17_FA_ST8_123232 | x | x | x | x | x | x | |||||||||
| Sau_17_MO_ST8_123221 | x | x | x | x | x | x | |||||||||
| Sau_17_DO_ST8_123220 | x | x | x | x | x | x | |||||||||
| Sau_18_GF_ST8_153686-7 | x | x | x | x | x | x | |||||||||
| Sau_18_GM_ST8_153686-6 | x | x | x | x | x | x | |||||||||
| Sau_18_MO_ST10_153686-3 | x | ||||||||||||||
| Sau_19_MO_ST10_150824 | x | x | x | ||||||||||||
| Sau_19_CH_ST10_150819 | x | ||||||||||||||
| Sau_17_MO_ST22_123223 | x | ||||||||||||||
| Sau_18_FA_ST22_153686-2 | x | ||||||||||||||
| Sau_19_FA_ST22_150811 | x | ||||||||||||||
| Sau_18_GM_ST59_153686-5 | x | ||||||||||||||
| Sau_19_GF_ST59_150803 | x | ||||||||||||||
| Sau_18_CH_ST707_153686-4 | x | ||||||||||||||
| Sau_18_DO_ST7204_153686-1 | x | ||||||||||||||
| Sca_19_DO_STX_150837 | x | ||||||||||||||
| Sep_19_GF_ST153_150806 | x | x | x | x | |||||||||||
| Sep_17_CH_ST1133_123222 | x | x | x | x | x | x | |||||||||
| Sep_19_GM_ST5_150797 | x | x | x | x | x | x | x | x | x | x | |||||
| Slu_19_GM_ST2_150795 | x | ||||||||||||||
| Sps_17_DO_ST2169_123218 | x | x | x | x | |||||||||||
| Sps_19_DO_ST2169_150832 | x | x | x | x | |||||||||||
| Sps_17_DO_ST2168_123216 | x | x | x | x | x | ||||||||||
| Sps_19_DO_ST2168_150831 | x | x | x | x | x | ||||||||||
| Swa_19_FA_STX_150815 |
| S. aureus | 69 | 11 | phi2958PVL | StauST398_2 | phiJB | phi2958PVL | P282 | YMC/09/04/R1988 |
|---|---|---|---|---|---|---|---|---|
| Sau_18_MO_ST10_153686-3 | x | x | ||||||
| Sau_19_MO_ST10_150824 | x | x | ||||||
| Sau_19_CH_ST10_150819 | x | x | ||||||
| Sau_18_CH_ST707_153686-4 | x | |||||||
| Sau_18_DO_ST7204_153686-1 | x | |||||||
| Sau_17_FA_ST8_123232 | ||||||||
| Sau_17_MO_ST8_123221 | x | x | ||||||
| Sau_17_DO_ST8_123220 | x | x | ||||||
| Sau_18_GM_ST8_153686-6 | x | x | ||||||
| Sau_18_GF_ST8_153686-7 | x | x | ||||||
| S. epidermidis | StB20_like | Ipla5 | ||||||
| Sep_17_CH_ST1133_123222 | ||||||||
| Sep_19_GF_ST153_150806 | x | x | ||||||
| Sep_19_GM_ST5_150797 |
| Sample | rep19 | rep39 | rep7a | repUS43 | rep7c | rep16 | repUS5 | rep20 | repUS35 | rep22 | repUS19 | repUS12 | rep5b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sau_17_DO_ST8_123220 | x | x | x | ||||||||||
| Sau_17_FA_ST8_123232 | x | x | x | x | |||||||||
| Sau_18_GF_ST8_153686-7 | x | x | x | ||||||||||
| Sau_18_GM_ST8_153686-6 | x | x | x | ||||||||||
| Sau_17_MO_ST8_123221 | x | x | x | ||||||||||
| Sau_18_CH_ST707_153686-4 | x | x | |||||||||||
| Sau_18_DO_ST7204_153686-1 | x | x | |||||||||||
| Sau_18_FA_ST22_153686-2 | |||||||||||||
| Sau_17_MO_ST22_123223 | |||||||||||||
| Sau_19_FA_ST22_150811 | |||||||||||||
| Sau_19_MO_ST10_150824 | x | x | |||||||||||
| Sau_18_MO_ST10_153686-3 | |||||||||||||
| Sau_19_CH_ST10_150819 | |||||||||||||
| Sau_19_GF_ST59_150803 | |||||||||||||
| Sau_18_GM_ST59_153686-5 | |||||||||||||
| Sca_19_DO_STX_150837 | x | ||||||||||||
| Sep_17_CH_ST1133_123222 | x | x | x | ||||||||||
| Sep_19_GF_ST153_150806 | x | ||||||||||||
| Sep_19_GM_ST5_150797 | x | x | x | x | |||||||||
| Slu_19_GM_ST2_150795 | x | ||||||||||||
| Sps_17_DO_ST2169_123218 | x | ||||||||||||
| Sps_17_DO_ST2168_123216 | x | ||||||||||||
| Sps_19_DO_ST2169_150832 | x | ||||||||||||
| Sps_19_DO_ST2168_150831 | x | ||||||||||||
| Swa_19_FA_STX_150815 | x |
| Sample | scn | sep | sea | sak | chp | IEC-Variant | Sample |
|---|---|---|---|---|---|---|---|
| Sau_17_DO_ST8_123220 | x | x | x | B | Sau_17_DO_ST8_123220 | ||
| Sau_17_FA_ST8_123232 | x | x | x | B | Sau_17_FA_ST8_123232 | ||
| Sau_18_GF_ST8_153686-7 | x | x | x | B | Sau_18_GF_ST8_153686-7 | ||
| Sau_18_GM_ST8_153686-6 | x | x | x | B | Sau_18_GM_ST8_153686-6 | ||
| Sau_17_MO_ST8_123221 | x | x | x | B | Sau_17_MO_ST8_123221 | ||
| Sau_18_CH_ST707_153686-4 | x | x | x | B | Sau_18_CH_ST707_153686-4 | ||
| Sau_18_DO_ST7204_153686-1 | x | x | x | B | Sau_18_DO_ST7204_153686-1 | ||
| Sau_18_FA_ST22_153686-2 | x | x | x | B | Sau_18_FA_ST22_153686-2 | ||
| Sau_17_MO_ST22_123223 | x | x | x | B | Sau_17_MO_ST22_123223 | ||
| Sau_19_FA_ST22_150811 | x | x | x | B | Sau_19_FA_ST22_150811 | ||
| Sau_19_MO_ST10_150824 | x | x | E | Sau_19_MO_ST10_150824 | |||
| Sau_18_MO_ST10_153686-3 | x | x | E | Sau_18_MO_ST10_153686-3 | |||
| Sau_19_CH_ST10_150819 | x | x | E | Sau_19_CH_ST10_150819 | |||
| Sau_19_GF_ST59_150803 | x | x | C | Sau_19_GF_ST59_150803 | |||
| Sau_18_GM_ST59_153686-5 | x | x | C | Sau_18_GM_ST59_153686-5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsini, M.; Petrin, S.; Corrò, M.; Baggio, G.; Spagnolo, E.; Losasso, C. Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study. Pathogens 2022, 11, 802. https://doi.org/10.3390/pathogens11070802
Orsini M, Petrin S, Corrò M, Baggio G, Spagnolo E, Losasso C. Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study. Pathogens. 2022; 11(7):802. https://doi.org/10.3390/pathogens11070802
Chicago/Turabian StyleOrsini, Massimiliano, Sara Petrin, Michela Corrò, Giulia Baggio, Elena Spagnolo, and Carmen Losasso. 2022. "Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study" Pathogens 11, no. 7: 802. https://doi.org/10.3390/pathogens11070802
APA StyleOrsini, M., Petrin, S., Corrò, M., Baggio, G., Spagnolo, E., & Losasso, C. (2022). Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study. Pathogens, 11(7), 802. https://doi.org/10.3390/pathogens11070802