
Establishing a Herpesvirus Quiescent Infection in Differentiated Human Dorsal Root Ganglion Neuronal Cell Line Mediated by Micro-RNA Overexpression


Abstract
:1. Introduction
2. Results
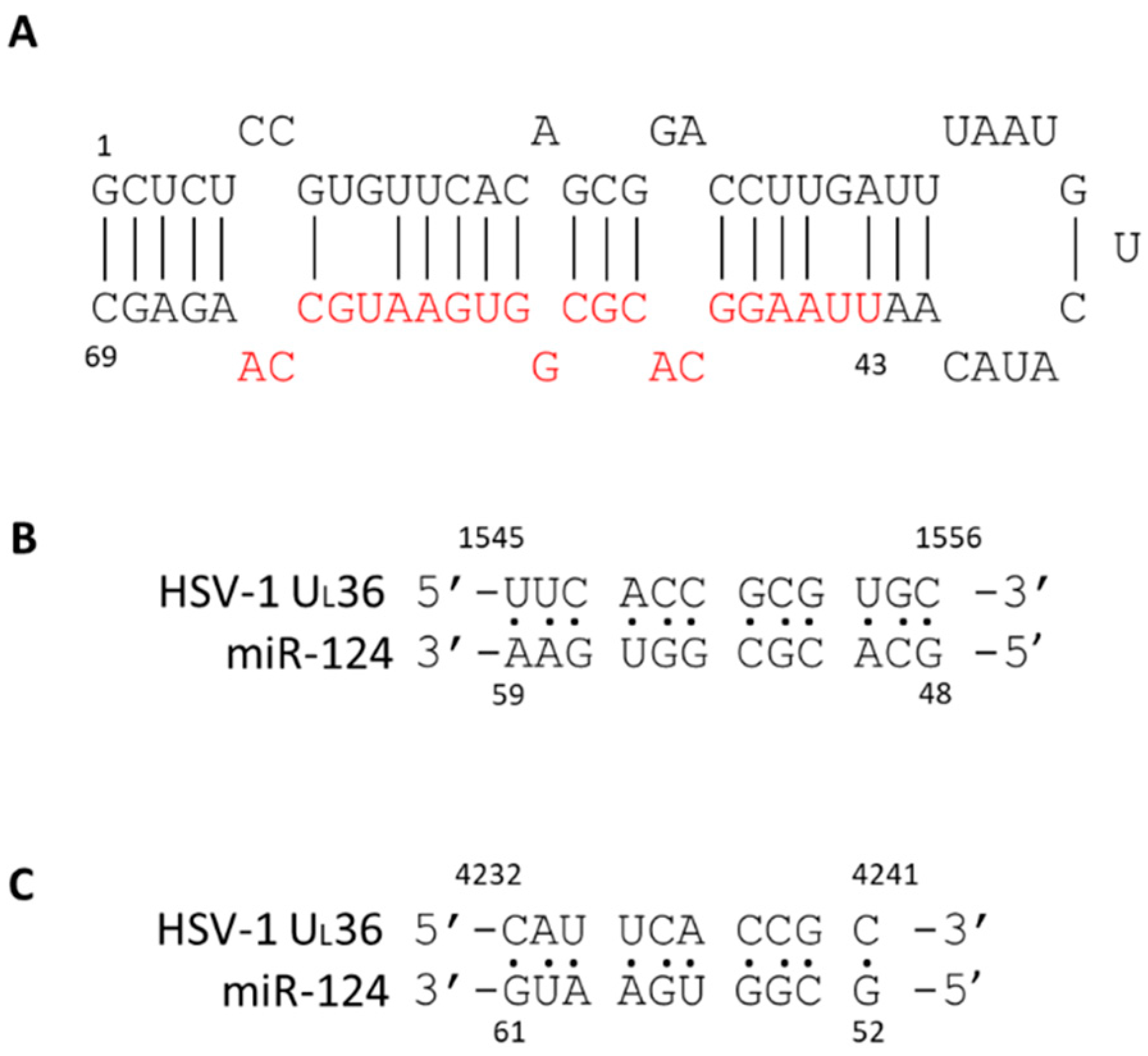
2.1. The miR124 Expression Increased in HD10.6 Cells
2.2. HSV-1 Can Infect Differentiated HD10 Cells with miR124 Overexpression
2.3. The miR124 Expression Reduced the Size of the HD10.6 Cells
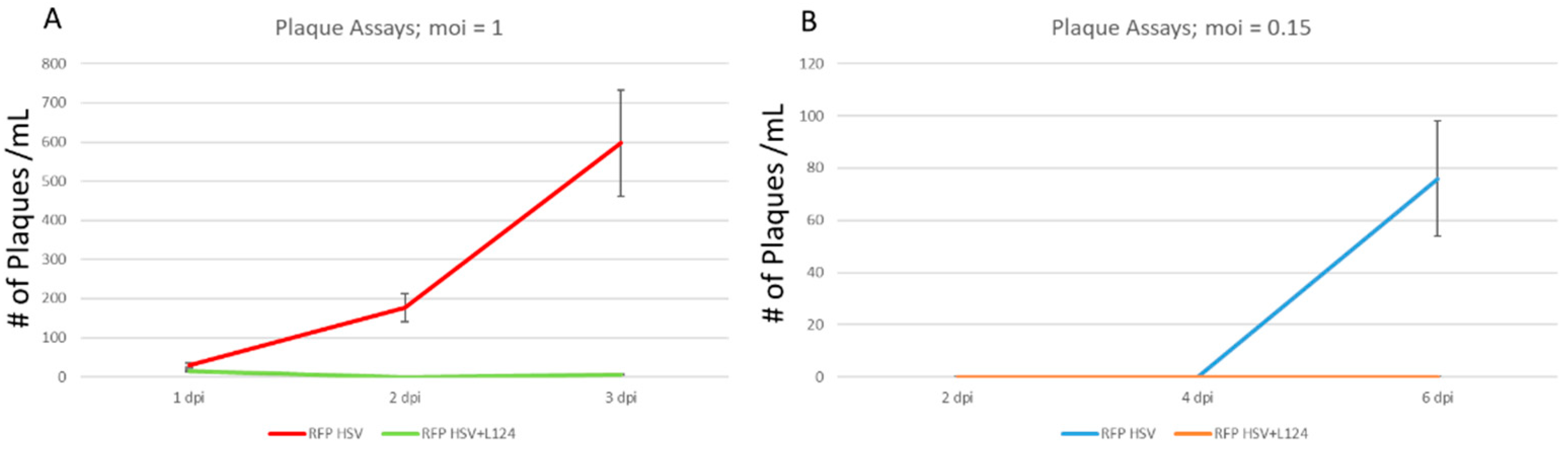
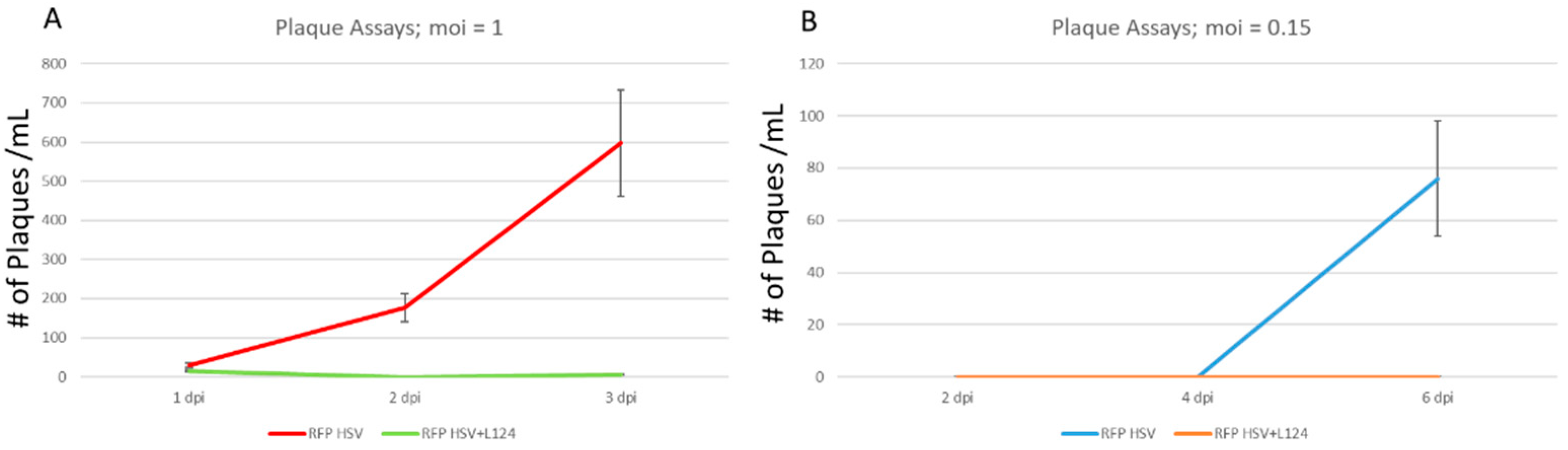
2.4. The Overexpression of miR124 Promoted a Quiescent State of HSV-1 Infection
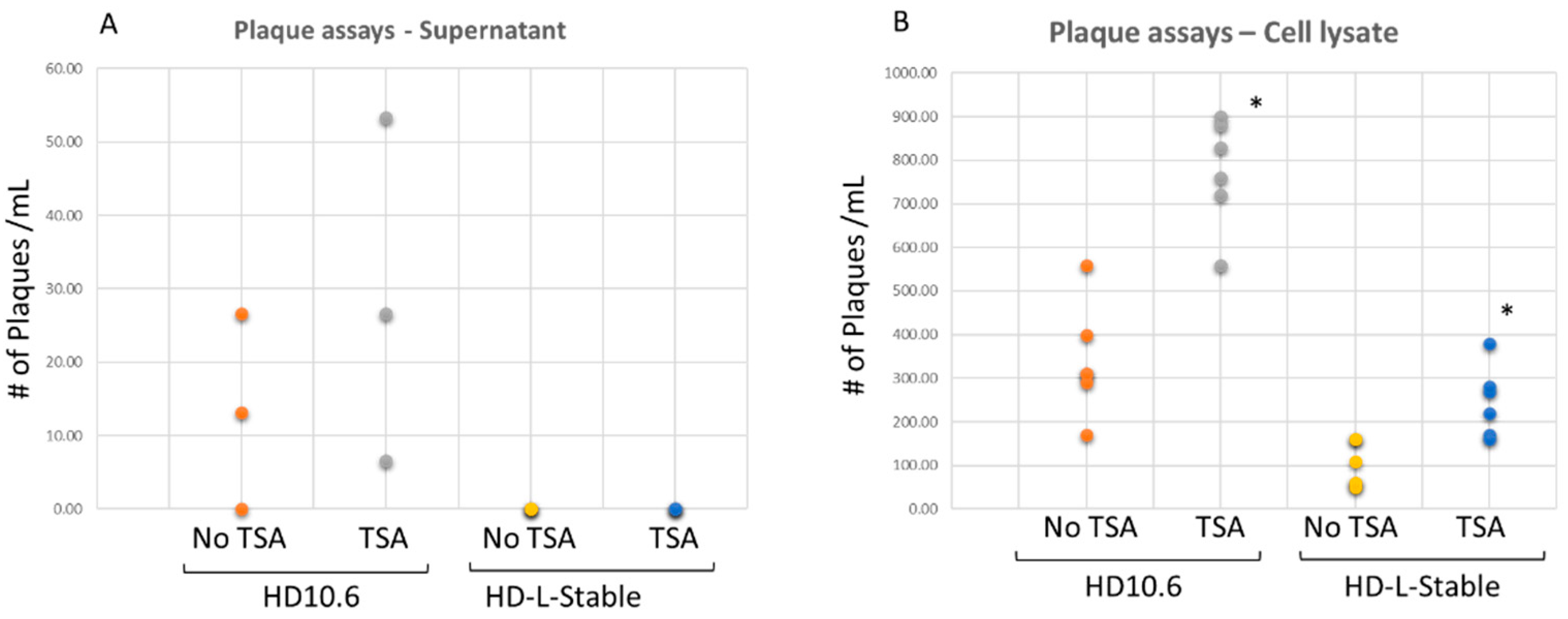
2.5. HSV-1 Replication Occurred but Failed to Be Released from the HD-L-Stable Cells
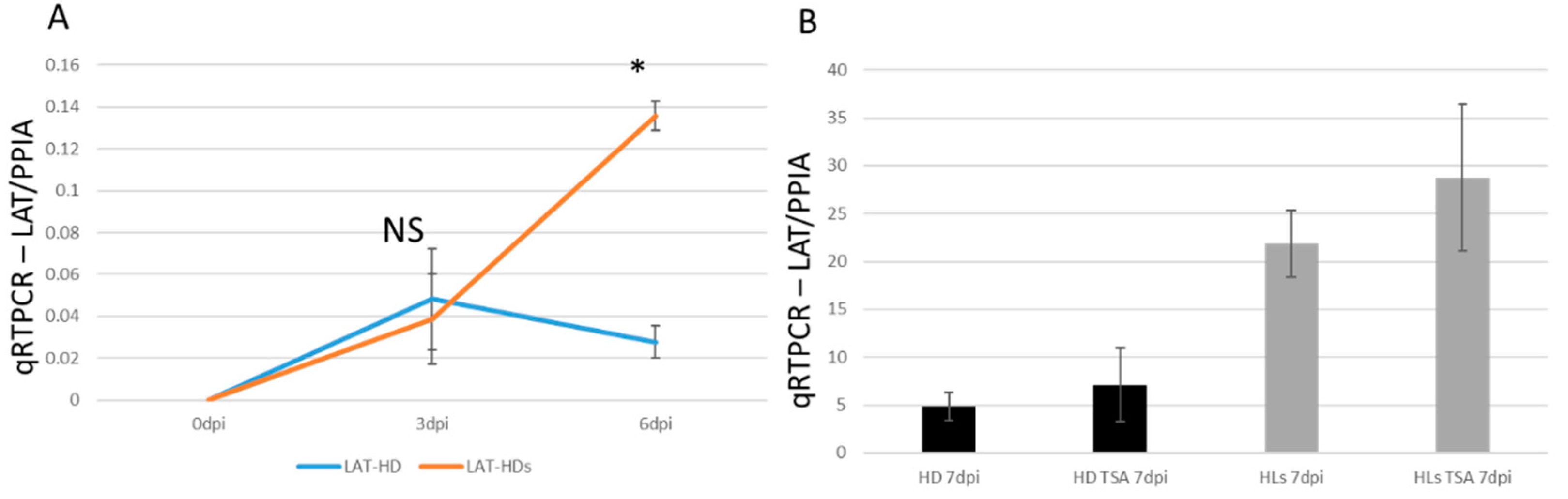
2.6. TSA Increased the Viral Gene Transcription from the Quiescent State of Infection
2.7. HSV-1 LAT Accumulated in the miR124 Expressing HD10.6 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Proliferation
4.2. Cell Differentiation
4.3. Introduction of miR-124 into HD10.6 Cells
4.4. Infection of HD10.6 by HSV-1
4.5. Electrophysiology
4.6. RNA Extraction and Reverse Transcription
4.7. Quantitative PCR (qPCR)
4.8. Plaque Assay
4.9. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitley, R.J. Antiviral treatment of a serious herpes simplex infection: Encephalitis. J Am. Acad Dermatol. 1988, 18 Pt 2, 209–211. [Google Scholar] [CrossRef]
- Canivet, C.; Menasria, R.; Rheaume, C.; Piret, J.; Boivin, G. Valacyclovir combined with artesunate or rapamycin improves the outcome of herpes simplex virus encephalitis in mice compared to antiviral therapy alone. Antivir. Res. 2015, 123, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, G.; Mishra, A.K.; Tandon, R.; Sharma, M.K.; Sharma, A.; Nayak, N.; Titiyal, J.S.; Sharma, N. Evaluation of tear samples for Herpes Simplex Virus 1 (HSV) detection in suspected cases of viral keratitis using PCR assay and conventional laboratory diagnostic tools. Br. J. Ophthalmol. 2011, 95, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramod, N.P.; Thyagarajan, S.P.; Mohan, K.V.; Anandakannan, K. Polymerase chain reaction in the diagnosis of herpetic keratitis: Experience in a developing country. Can. J. Ophthalmol. J. Can. D’ophtalmologie. 2000, 35, 134–140. [Google Scholar] [CrossRef]
- Jabs, D.A. Acyclovir for recurrent herpes simplex virus ocular disease. N. Engl. J. Med. 1998, 339, 340–341. [Google Scholar] [CrossRef]
- Kutsche, L.K.; Gysi, D.M.; Fallmann, J.; Lenk, K.; Petri, R.; Swiersy, A.; Klapper, S.D.; Pircs, K.; Khattak, S.; Stadler, P.F.; et al. Combined Experimental and System-Level Analyses Reveal the Complex Regulatory Network of miR-124 during Human Neurogenesis. Cell Syst. 2018, 7, 438–452.e8. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. CB 2002, 12, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [Green Version]
- Hou, Q.; Ruan, H.; Gilbert, J.; Wang, G.; Ma, Q.; Yao, W.D.; Man, H.Y. MicroRNA miR124 is required for the expression of homeostatic synaptic plasticity. Nat. Commun. 2015, 6, 10045. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; Chomyk, A.M.; Chang, A.; Ribaudo, M.V.; Deckard, S.A.; Doud, M.K.; Edberg, D.D.; Bai, B.; Li, M.; Baranzini, S.E.; et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann. Neurol. 2013, 73, 637–645. [Google Scholar] [CrossRef]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-associated microRNA-1 and -133b suppress the Warburg effect in colorectal tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzacurati, L.; Marzulli, M.; Reinhart, B.; Miyagawa, Y.; Uchida, H.; Goins, W.F.; Li, A.; Kaur, B.; Caligiuri, M.; Cripe, T.; et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol. J. Am. Soc. Gene Ther. 2015, 23, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Gao, Y.; Zhou, Q.; Zhang, Q.; Peng, Y.; Tian, K.; Wang, J.; Zheng, X. Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene 2014, 536, 272–278. [Google Scholar] [CrossRef]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- D’Aiuto, L.; Bloom, D.C.; Naciri, J.N.; Smith, A.; Edwards, T.G.; McClain, L.; Callio, J.A.; Jessup, M.; Wood, J.; Chowdari, K.; et al. Modeling Herpes Simplex Virus 1 Infections in Human Central Nervous System Neuronal Cells Using Two- and Three-Dimensional Cultures Derived from Induced Pluripotent Stem Cells. J. Virol. 2019, 93, e00111-19. [Google Scholar] [CrossRef] [Green Version]
- Edwards, T.G.; Bloom, D.C. Lund Human Mesencephalic (LUHMES) Neuronal Cell Line Supports Herpes Simplex Virus 1 Latency In Vitro. J Virol. 2019, 93, e02210-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Martin-Caraballo, M.; Hsia, S.V. Modulation of Voltage-Gated Sodium Channel Activity in Human Dorsal Root Ganglion Neurons by Herpesvirus Quiescent Infection. J. Virol. 2020, 94, e01823-19. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [Green Version]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Thellman, N.M.; Botting, C.; Madaj, Z.; Triezenberg, S.J. An Immortalized Human Dorsal Root Ganglion Cell Line Provides a Novel Context To Study Herpes Simplex Virus 1 Latency and Reactivation. J. Virol. 2017, 91, e00080-17. [Google Scholar] [CrossRef] [Green Version]
- Thellman, N.M.; Triezenberg, S.J. Herpes Simplex Virus Establishment, Maintenance, and Reactivation: In Vitro Modeling of Latency. Pathogens 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv. Virus Res. 2016, 94, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Pires de Mello, C.P.; Bloom, D.C.; Paixao, I.C. Herpes simplex virus type-1: Replication, latency, reactivation, and its antiviral targets. Antivir. Ther. 2016, 21, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesola, J.M.; Zhu, J.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J. Virol. 2005, 79, 14516–14525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourchet, A.; Modrek, A.S.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ljubimov, A.V.; Jin, L.; Pfeffer, K.; Kronenberg, M.; Ghiasi, H. Herpes Simplex Virus 1 Latency and the Kinetics of Reactivation Are Regulated by a Complex Network of Interactions between the Herpesvirus Entry Mediator, Its Ligands (gD, BTLA, LIGHT, and CD160), and the Latency-Associated Transcript. J. Virol. 2018, 92, e01451-18. [Google Scholar] [CrossRef] [Green Version]
- Sawtell, N.M.; Thompson, R.L. Herpes simplex virus and the lexicon of latency and reactivation: A call for defining terms and building an integrated collective framework. F1000Research 2016, 5, e01451-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Mahalingam, G.; Imai, Y.; Pesnicak, L.; Margolis, T.P.; Straus, S.E.; Cohen, J.I. Cell type-specific accumulation of the major latency-associated transcript (LAT) of herpes simplex virus type 2 in LAT transgenic mice. Virology 2009, 386, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.L.; Preston, C.M.; Sawtell, N.M. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sawtell, N.M. The herpes simplex virus type 1 latency-associated transcript locus is required for the maintenance of reactivation competent latent infections. J. Neurovirol. 2011, 17, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Spivack, J.G.; Fraser, N.W. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol. 1987, 61, 3841–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Kubat, N.J.; Amelio, A.L.; Giordani, N.V.; Bloom, D.C. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J. Virol. 2004, 78, 12508–12518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcox, C.L.; Johnson, E.M., Jr. Characterization of nerve growth factor-dependent herpes simplex virus latency in neurons in vitro. J. Virol. 1988, 62, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Bertke, A.S.; Swanson, S.M.; Chen, J.; Imai, Y.; Kinchington, P.R.; Margolis, T.P. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 2011, 85, 6669–6677. [Google Scholar] [CrossRef] [Green Version]
- Raymon, H.K.; Thode, S.; Zhou, J.; Friedman, G.C.; Pardinas, J.R.; Barrere, C.; Johnson, R.M.; Sah, D.W. Immortalized human dorsal root ganglion cells differentiate into neurons with nociceptive properties. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 5420–5428. [Google Scholar] [CrossRef] [Green Version]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell. 2007, 27, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P.; Chen, C.Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004, 5, 396–400. [Google Scholar] [CrossRef]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Darnell, D.K.; Kaur, S.; Stanislaw, S.; Konieczka, J.H.; Yatskievych, T.A.; Antin, P.B. MicroRNA expression during chick embryo development. Dev. Dyn. 2006, 235, 3156–3165. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.A.; Tapia-Ramirez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunyak, V.V.; Burgess, R.; Prefontaine, G.G.; Nelson, C.; Sze, S.H.; Chenoweth, J.; Schwartz, P.; Pevzner, P.A.; Glass, C.; Mandel, G.; et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 2002, 298, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Ballas, N.; Mandel, G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr. Opin. Neurobiol. 2005, 15, 500–506. [Google Scholar] [CrossRef]
- Du, T.; Zhou, G.; Khan, S.; Gu, H.; Roizman, B. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc. Natl. Acad. Sci. USA 2010, 107, 15904–15909. [Google Scholar] [CrossRef] [Green Version]
- Conaco, C.; Otto, S.; Han, J.J.; Mandel, G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl. Acad. Sci. USA 2006, 103, 2422–2427. [Google Scholar] [CrossRef] [Green Version]
- Yeo, M.; Lee, S.K.; Lee, B.; Ruiz, E.C.; Pfaff, S.L.; Gill, G.N. Small CTD phosphatases function in silencing neuronal gene expression. Science 2005, 307, 596–600. [Google Scholar] [CrossRef]
- Visvanathan, J.; Lee, S.; Lee, B.; Lee, J.W.; Lee, S.K. The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 2007, 21, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Du, T.; Roizman, B. The role of the CoREST/REST repressor complex in herpes simplex virus 1 productive infection and in latency. Viruses 2013, 5, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Pinnoji, R.C.; Bedadala, G.R.; George, B.; Holland, T.C.; Hill, J.M.; Hsia, S.C. Repressor element-1 silencing transcription factor/neuronal restrictive silencer factor (REST/NRSF) can regulate HSV-1 immediate-early transcription via histone modification. Virol. J. 2007, 4, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Pfaff, S.L.; Gage, F.H. A functional study of miR-124 in the developing neural tube. Genes Dev. 2007, 21, 531–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottcher, S.; Maresch, C.; Granzow, H.; Klupp, B.G.; Teifke, J.P.; Mettenleiter, T.C. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J. Virol. 2008, 82, 6009–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffmaster, N.J.; Sollars, P.J.; Richards, A.L.; Pickard, G.E.; Smith, G.A. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc. Natl. Acad. Sci. USA 2015, 112, 12818–12823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.; Sexton, G.L.; Huang, E.; Person, S. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J. Virol. 2008, 82, 11354–11361. [Google Scholar] [CrossRef] [Green Version]
- Pachuau, J.; Martin-Caraballo, M. Extrinsic regulation of T-type Ca(2+) channel expression in chick nodose ganglion neurons. Dev. Neurobiol. 2007, 67, 1915–1931. [Google Scholar] [CrossRef] [Green Version]
- Pachuau, J.; Martin-Caraballo, M. Expression pattern of T-type Ca(2+) channels in embryonic chick nodose ganglion neurons. Dev. Neurobiol. 2007, 67, 1901–1914. [Google Scholar] [CrossRef]
- Figliozzi, R.W.; Chen, F.; Chi, A.; Hsia, S.C. Using the inverse Poisson distribution to calculate multiplicity of infection and viral replication by a high-throughput fluorescent imaging system. Virol. Sinica. 2016, 31, 180–183. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target [gene] | Primer Sequence | Annealing Temp |
|---|---|---|
| miR-26a | CGAGTTCAAGTAATCCAGGA (f); CCAGTTTTTTTTTTTTTTTAGCCTATC (r) | 60 °C |
| miR-124 | TAAGGCACGCGGTGA (f); CCAGTTTTTTTTTTTTTTTGGCAT (r) | 60 °C |
| PPIA(h] | AGCATACGGGTCCTGGCATCT(f); CATGCTTGCCATCCAACCACTCA(r) | 65 °C |
| GFP | ACTTCAAGATCCGCCACAACA (f); TGATCGCGCTTCTCGTTGG (r) | 58 °C |
| ICP0 (v) | GACGGGCAATCAGCGGTTC(f); GTAGTCTGCGTCGTCCAGGT(r) | 60 °C |
| TK (v) | TACCCGAGCCGATGACTTAC(f); AAGGCATGCCCATTGTTATC(r) | 62 °C |
| LAT (v) | CGGCGACATCCTCCCCCTAAGC(f); GACAGACGAACGAAACGTTCCG(r) | 60 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-C.; Li, H.; Martin-Caraballo, M.; Hsia, S.V. Establishing a Herpesvirus Quiescent Infection in Differentiated Human Dorsal Root Ganglion Neuronal Cell Line Mediated by Micro-RNA Overexpression. Pathogens 2022, 11, 803. https://doi.org/10.3390/pathogens11070803
Chen Y-C, Li H, Martin-Caraballo M, Hsia SV. Establishing a Herpesvirus Quiescent Infection in Differentiated Human Dorsal Root Ganglion Neuronal Cell Line Mediated by Micro-RNA Overexpression. Pathogens. 2022; 11(7):803. https://doi.org/10.3390/pathogens11070803
Chicago/Turabian StyleChen, Yu-Chih, Hedong Li, Miguel Martin-Caraballo, and Shaochung Victor Hsia. 2022. "Establishing a Herpesvirus Quiescent Infection in Differentiated Human Dorsal Root Ganglion Neuronal Cell Line Mediated by Micro-RNA Overexpression" Pathogens 11, no. 7: 803. https://doi.org/10.3390/pathogens11070803
APA StyleChen, Y.-C., Li, H., Martin-Caraballo, M., & Hsia, S. V. (2022). Establishing a Herpesvirus Quiescent Infection in Differentiated Human Dorsal Root Ganglion Neuronal Cell Line Mediated by Micro-RNA Overexpression. Pathogens, 11(7), 803. https://doi.org/10.3390/pathogens11070803