
Legionella pneumophila Infection of Human Macrophages Retains Golgi Structure but Reduces O-Glycans

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. L. pneumophila Growth and Cell Infections and Treatments
2.4. Immunostaining and Imaging
2.5. Immunoblotting
2.6. Data Analysis and Statistics
3. Results
3.1. Golgi Morphology and Golgi Size Remains Intact in L. pneumophila-Infected U937 Human Macrophages
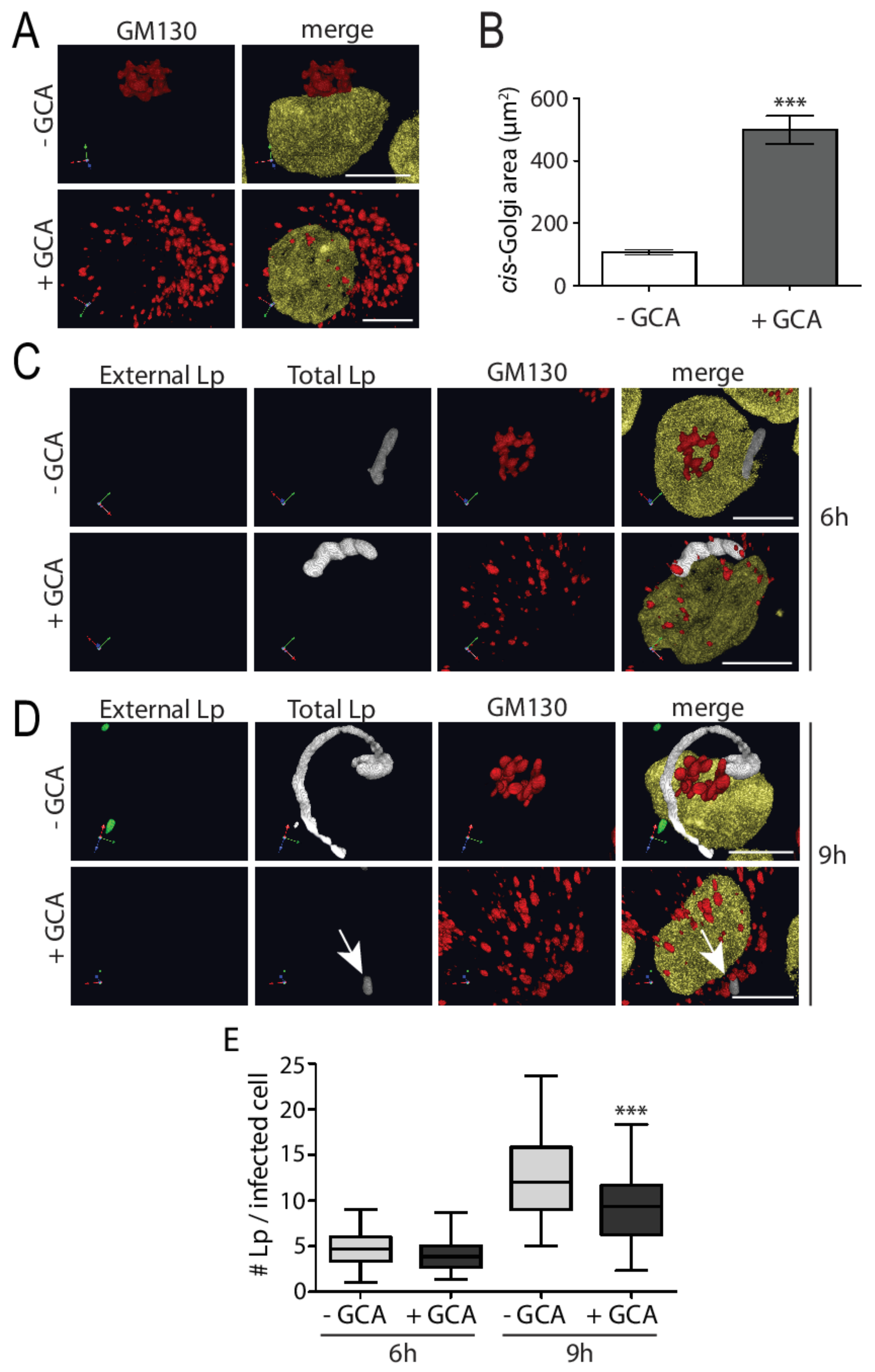
3.2. Experimental Golgi Fragmentation Decreases L. pneumophila Growth in Human Macrophages
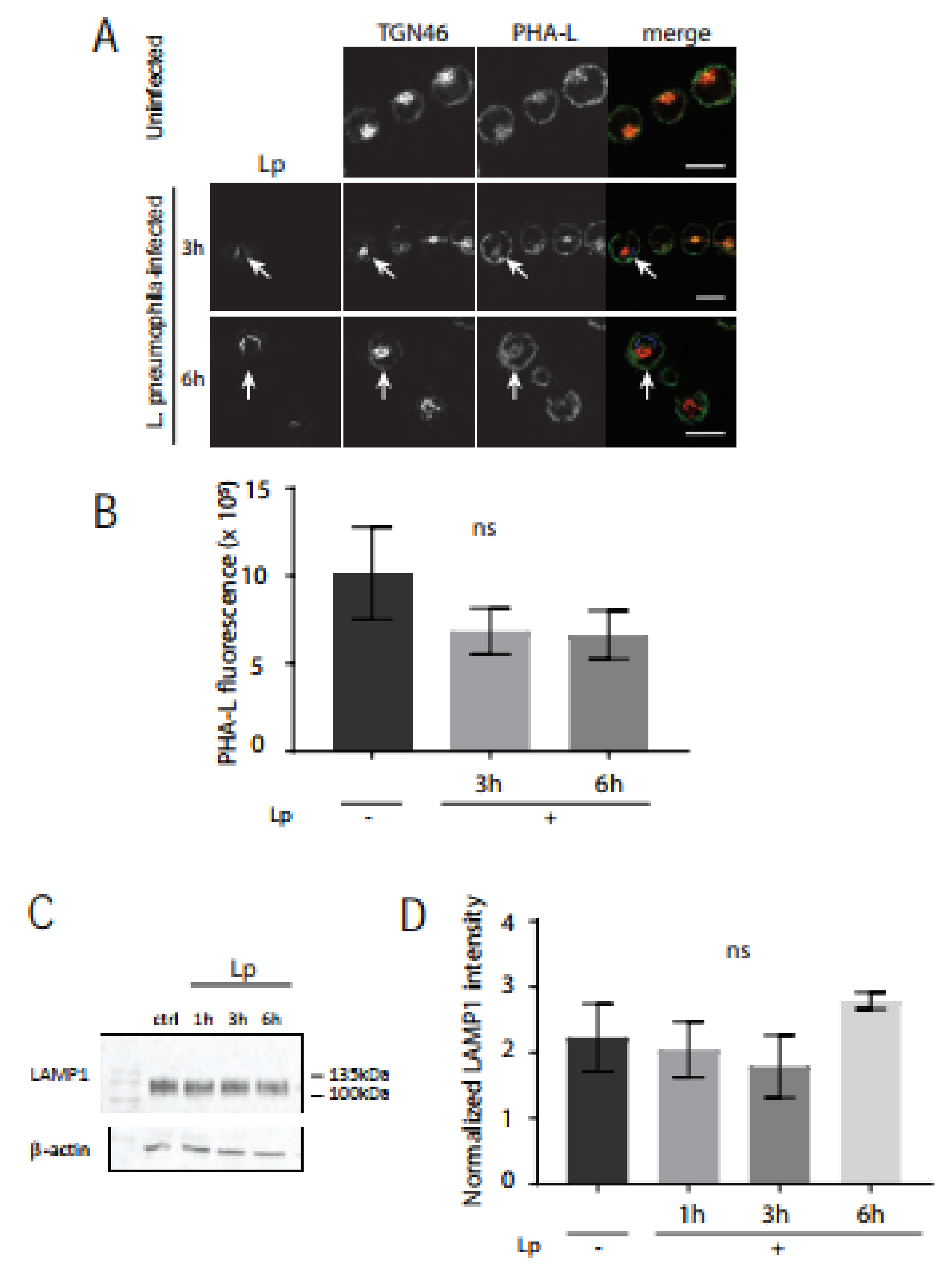
3.3. L. pneumophila Infection of Macrophages Decreases Lectin Intensity for O-Glycans but Not N-Glycans
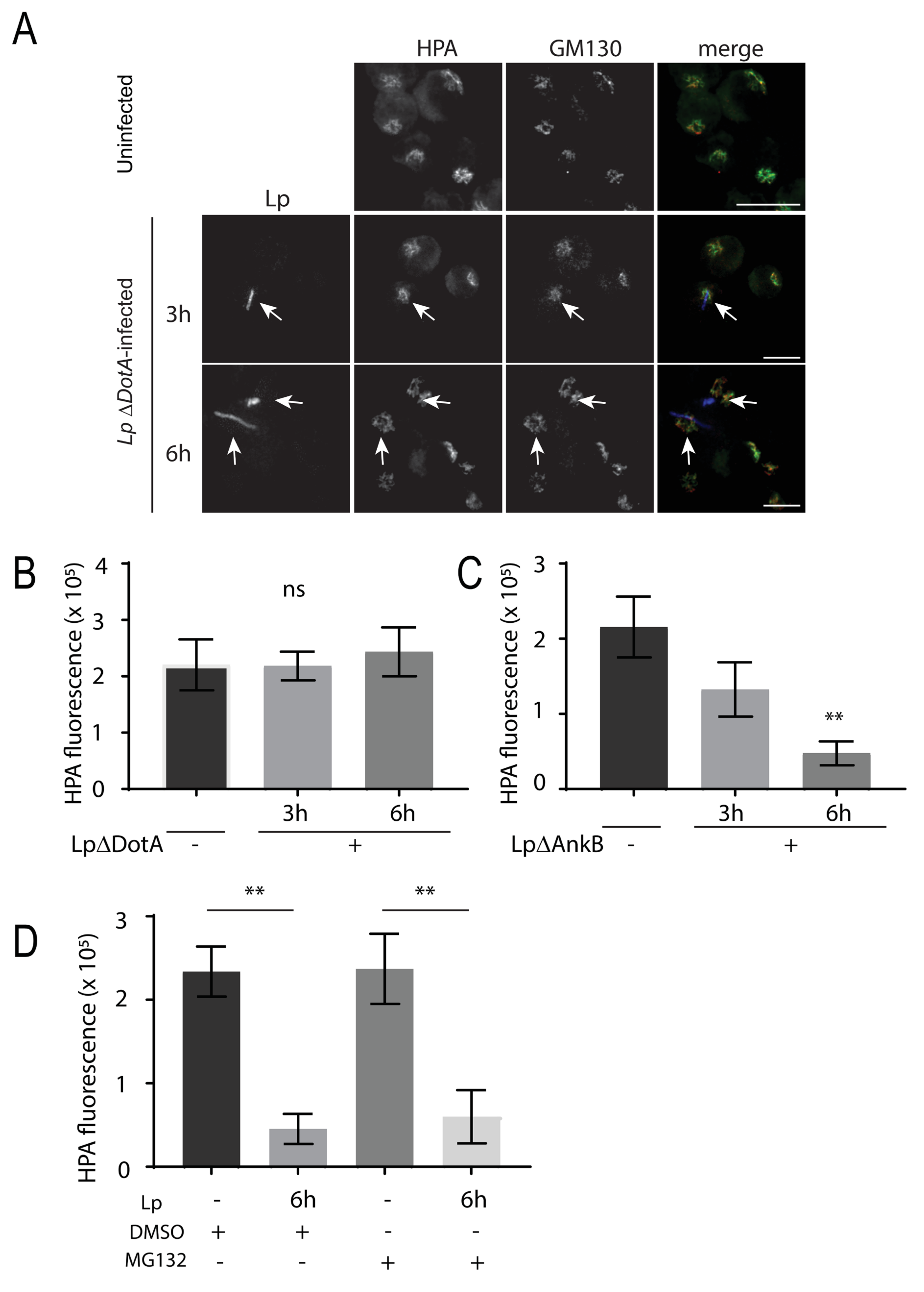
3.4. SLC35A2 Transporters Are Significantly Reduced in L. pneumophila-Infected Macrophages and GalNAc Supplementation Supports Bacteria Growth
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierre, D.M.; Baron, J.; Yu, V.L.; Stout, J.E. Diagnostic Testing for Legionnaires’ Disease. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Cunha, B.A.; Burillo, A.; Bouza, E. Legionnaires’ Disease. Lancet 2016, 387, 376–385. [Google Scholar] [CrossRef]
- Ensminger, A.W.; Isberg, R.R. Legionella Pneumophila Dot/Icm Translocated Substrates: A Sum of Parts. Curr. Opin. Microbiol. 2009, 12, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Zink, S.D.; Pedersen, L.; Cianciotto, N.P.; Abu Kwaik, Y. The Dot/Icm Type IV Secretion System of Legionella Pneumophila Is Essential for the Induction of Apoptosis in Human Macrophages. Infect. Immun. 2002, 70, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.R.; Tilney, L.G. The Road Less Traveled Transport of Legionella to the Endoplasmic Reticulum. J. Cell Biol. 2002, 158, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Stein, M.P.; Pypaert, M.; Roy, C.R. Legionella Subvert the Functions of Rab1 and Sec22b to Create a Replicative Organelle. J. Exp. Med. 2004, 199, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, M.; Matsuo, H.; Koito, T.; Murata, M.; Kubori, T.; Nagai, H.; Tagaya, M.; Arasaki, K. Legionella Hijacks the Host Golgi-to-ER Retrograde Pathway for the Association of Legionella-Containing Vacuole with the ER. PLoS Pathog. 2021, 17, e1009437. [Google Scholar] [CrossRef]
- Brombacher, E.; Urwyler, S.; Ragaz, C.; Weber, S.S.; Kami, K.; Overduin, M.; Hilbi, H. Rab1 Guanine Nucleotide Exchange Factor SidM Is a Major Phosphatidylinositol 4-Phosphate-Binding Effector Protein of Legionella Pneumophila. J. Biol. Chem. 2009, 284, 4846–4856. [Google Scholar] [CrossRef]
- Machner, M.P.; Isberg, R.R. Targeting of Host Rab GTPase Function by the Intravacuolar Pathogen Legionella Pneumophila. Dev. Cell 2006, 11, 47–56. [Google Scholar] [CrossRef]
- Machner, M.; Isberg, R. A Bifunctional Bacterial Protein Links GDI Displacement of Rab1 Activation. Science 2007, 318, 974–977. [Google Scholar] [CrossRef]
- Hardiman, C.A.; Roy, C.R. AMPylation Is Critical for Rab1 Localization to Vacuoles Containing Legionella Pneumophila. mBio 2014, 5, e01035-13. [Google Scholar] [CrossRef]
- Müller, M.P.; Peters, H.; Blümer, J.; Blankenfeldt, W.; Goody, R.S.; Itzen, A. The Legionella Effector Protein DrrA AMPylates the Membrane Traffic Regulator Rab1b. Science 2010, 329, 946–949. [Google Scholar] [CrossRef]
- Ingmundson, A.; Delprato, A.; Lambright, D.; Roy, C. Legionella Pneumophila Proteins That Regulate Rab1 Membrane Cycling. Nature 2007, 450, 365–369. [Google Scholar] [CrossRef]
- Goody, P.R.; Heller, K.; Oesterlin, L.K.; Muller, M.P.; Itzen, A.; Goody, R.S. Reversible Phosphocholination of Rab Proteins by Legionella Pneumophila Effector Proteins. EMBO J. 2012, 31, 1774–1784. [Google Scholar] [CrossRef]
- Mukherjee, S.; Liu, X.; Arasaki, K.; McDonough, J.; Galán, J.E.; Roy, C.R. Modulation of Rab GTPase Function by a Protein Phosphocholine Transferase. Nature 2011, 477, 103–106. [Google Scholar] [CrossRef]
- Nagai, J.; Kagan, J.; Zhu, X.; Kahn, R.; Roy, C. A Bacterial Guanine Nucleotide Exchange Factor Activates ARF on Legionella Phagosomes. Science 2002, 295, 679–682. [Google Scholar] [CrossRef]
- Alix, E.; Chesnel, L.; Bowzard, B.J.; Tucker, A.M.; Delprato, A.; Cherfils, J.; Wood, D.O.; Kahn, R.A.; Roy, C.R. The Capping Domain in RalF Regulates Effector Functions. PLoS Pathog. 2012, 8, e1003012. [Google Scholar] [CrossRef]
- Robinson, C.G.; Roy, C.R. Attachment and Fusion of Endoplasmic Reticulum with Vacuoles Containing Legionella Pneumophila. Cell. Microbiol. 2006, 8, 793–805. [Google Scholar] [CrossRef]
- Kotewicz, K.M.; Ramabhadran, V.; Sjoblom, N.; Vogel, J.P.; Haenssler, E.; Zhang, M.; Behringer, J.; Scheck, R.A.; Isberg, R.R. A Single Legionella Effector Catalyzes a Multi-Step Ubiquitination Pathway to Rearrange Tubular Endoplasmic Reticulum for Replication. Cell Host Microbe 2017, 21, 169. [Google Scholar] [CrossRef]
- Li, J.; Ahat, E.; Wang, Y. Golgi Structure and Function in Health, Stress, and Diseases. Golgi Appar. Cent. 2019, 67, 441. [Google Scholar] [CrossRef]
- Stalder, D.; Gershlick, D.C. Direct Trafficking Pathways from the Golgi Apparatus to the Plasma Membrane. Semin. Cell Dev. Biol. 2020, 107, 112–125. [Google Scholar] [CrossRef]
- Li, Y.; Tran, A.H.; Danishefsky, S.J.; Tan, Z. Chemical Biology of Glycoproteins: From Chemical Synthesis to Biological Impact. Methods Enzymol. 2019, 621, 213–229. [Google Scholar] [CrossRef]
- van den Steen, P.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Concepts and Principles of O-Linked Glycosylation. Crit. Rev. Biochem. Mol. Biol. 2008, 33, 151–208. [Google Scholar] [CrossRef]
- Prashar, A.; Ortiz, M.E.; Lucarelli, S.; Barker, E.; Tabatabeiyazdi, Z.; Shamoun, F.; Raju, D.; Antonescu, C.; Guyard, C.; Terebiznik, M.R. Small Rho GTPases and the Effector VipA Mediate the Invasion of Epithelial Cells by Filamentous Legionella Pneumophila. Front. Cell. Infect. Microbiol. 2018, 8, 133. [Google Scholar] [CrossRef]
- Berger, K.H.; Isberg, R.R. Two Distinct Defects in Intracellular Growth Complemented by a Single Genetic Locus in Legionella Pneumophila. Mol. Microbiol. 1993, 7, 7–19. [Google Scholar] [CrossRef]
- Berger, K.H.; Merriam, J.J.; Isberg, R.R. Altered Intracellular Targeting Properties Associated with Mutations in the Legionella Pneumophila DotA Gene. Mol. Microbiol. 1994, 14, 809–822. [Google Scholar] [CrossRef]
- Ensminger, A.W.; Isberg, R.R. E3 Ubiquitin Ligase Activity and Targeting of BAT3 by Multiple Legionella Pneumophila Translocated Substrates. Infect. Immun. 2010, 78, 3905. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.S.; Sin, A.T.-W.; Poirier, M.B.; Harrison, R.E. Chlamydia Trachomatis Inclusion Disrupts Host Cell Cytokinesis to Enhance Its Growth in Multinuclear Cells. J. Cell. Biochem. 2016, 117, 132–143. [Google Scholar] [CrossRef]
- Sin, A.T.-W.; Harrison, R.E. Growth of the Mammalian Golgi Apparatus during Interphase. Mol. Cell. Biol. 2016, 36, 2344. [Google Scholar] [CrossRef]
- McCrossan, M.; Windsor, M.; Ponnambalam, S.; Armstrong, J.; Wileman, T. The Trans Golgi Network Is Lost from Cells Infected with African Swine Fever Virus. J. Virol. 2001, 75, 11755–11765. [Google Scholar] [CrossRef]
- Sáenz, J.B.; Sun, W.J.; Chang, J.W.; Li, J.; Bursulaya, B.; Gray, N.S.; Haslam, D.B. Golgicide A Reveals Essential Roles for GBF1 in Golgi Assembly and Function. Nat. Chem. Biol. 2009, 5, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Faucher, S.P.; Mueller, C.A.; Shuman, H.A. Legionella Pneumophila Transcriptome during Intracellular Multiplication in Human Macrophages. Front. Microbiol. 2011, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K. Intracellular Lectins Are Involved in Quality Control of Glycoproteins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 67–82. [Google Scholar] [CrossRef]
- Mitchell, B.S.; Schumacher, U. Histopathology Invited Re Vie W The Use of the Lectin Helixpomatia Agglutinin (HPA) as a Prognostic Indicator and as a Tool in Cancer Research. Histol. Histopathol. 1999, 14, 217–226. [Google Scholar]
- Hammarstrom, S.; Hammarstrom, M.L.; Sundblad, G.; Arnarp, J.; Lönngren, J. Mitogenic Leukoagglutinin from Phaseolus Vulgaris Binds to a Pentasaccharide Unit in N-Acetyllactosamine-Type Glycoprotein Glycans. Proc. Natl. Acad. Sci. USA 1982, 79, 1611–1615. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in Lysosome Biogenesis and Autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Song, Z. Roles of the Nucleotide Sugar Transporters (SLC35 Family) in Health and Disease. Mol. Asp. Med. 2013, 34, 590–600. [Google Scholar] [CrossRef]
- Hadley, B.; Litfin, T.; Day, C.J.; Haselhorst, T.; Zhou, Y.; Tiralongo, J. Nucleotide Sugar Transporter SLC35 Family Structure and Function. Comput. Struct. Biotechnol. J. 2019, 17, 1123–1134. [Google Scholar] [CrossRef]
- Tsuji, Y. Transmembrane Protein Western Blotting: Impact of Sample Preparation on Detection of SLC11A2 (DMT1) and SLC40A1 (Ferroportin). PLoS ONE 2020, 15, e0235563. [Google Scholar] [CrossRef]
- Nagai, H.; Roy, C.R. The DotA Protein from Legionella Pneumophila Is Secreted by a Novel Process That Requires the Dot/Icm Transporter. EMBO J. 2001, 20, 5962. [Google Scholar] [CrossRef]
- Price, C.T.D.; Al-Quadan, T.; Santic, M.; Rosenshine, I.; Abu Kwaik, Y. Host Proteasomal Degradation Generates Amino Acids Essential for Intracellular Bacterial Growth. Science 2011, 334, 1553–1557. [Google Scholar] [CrossRef]
- Qiu, J.; Luo, Z.Q. Hijacking of the Host Ubiquitin Network by Legionella Pneumophila. Front. Cell. Infect. Microbiol. 2017, 7, 487. [Google Scholar] [CrossRef]
- Liu, Y.; Mukherjee, R.; Bonn, F.; Colby, T.; Matic, I.; Glogger, M.; Heilemann, M.; Dikic, I. Serine-Ubiquitination Regulates Golgi Morphology and the Secretory Pathway upon Legionella Infection. Cell Death Differ. 2021, 28, 2957–2969. [Google Scholar] [CrossRef]
- Truchan, H.K.; Viebrock, L.; Cockburn, C.L.; Ojogun, N.; Griffin, B.P.; Wijesinghe, D.S.; Chalfant, C.E.; Carlyon, J.A. Anaplasma Phagocytophilum Rab10-Dependent Parasitism of the Trans-Golgi Network Is Critical for Completion of the Infection Cycle. Cell. Microbiol. 2016, 18, 260–281. [Google Scholar] [CrossRef]
- Beyer, A.R.; Rodino, K.G.; VieBrock, L.; Green, R.S.; Tegels, B.K.; Oliver, L.D.; Marconi, R.T.; Carlyon, J.A. Orientia Tsutsugamushi Ank9 Is a Multifunctional Effector That Utilizes a Novel GRIP-like Golgi Localization Domain for Golgi-to-Endoplasmic Reticulum Trafficking and Interacts with Host COPB2. Cell. Microbiol. 2017, 19, e12727. [Google Scholar] [CrossRef]
- Pokrovskaya, I.D.; Szwedo, J.W.; Goodwin, A.; Lupashina, T.V.; Nagarajan, U.M.; Lupashin, V.V. Chlamydia Trachomatis Hijacks Intra-Golgi COG Complex-Dependent Vesicle Trafficking Pathway. Cell. Microbiol. 2012, 14, 656–668. [Google Scholar] [CrossRef]
- Burnaevskiy, N.; Fox, T.G.; Plymire, D.A.; Ertelt, J.M.; Weigele, B.A.; Selyunin, A.S.; Way, S.S.; Patrie, S.M.; Alto, N.M. Proteolytic Elimination of N-Myristoyl Modifications by the Shigella Virulence Factor IpaJ. Nature 2013, 496, 106–109. [Google Scholar] [CrossRef]
- Mounier, J.; Boncompain, G.; Senerovic, L.; Lagache, T.; Chrétien, F.; Perez, F.; Kolbe, M.; Olivo-Marin, J.-C.; Sansonetti, P.J.; Sauvonnet, N. Shigella Effector IpaB-Induced Cholesterol Relocation Disrupts the Golgi Complex and Recycling Network to Inhibit Host Cell Secretion. Cell Host Microbe 2012, 12, 381–389. [Google Scholar] [CrossRef]
- Weber, S.; Ragaz, C.; Reus, K.; Nyfeler, Y.; Hilbi, H. Legionella Pneumophila Exploits PI(4)P to Anchor Secreted Proteins to the Replicative Vacuole. PLoS Pathog. 2006, 2, e46. [Google Scholar] [CrossRef]
- Ragaz, C.; Pietsch, H.; Urwyler, S.; Tiaden, A.; Weber, S.S.; Hilbi, H. The Legionella Pneumophila Phosphatidylinositol-4 Phosphate-Binding Type IV Substrate SidC Recruits Endoplasmic Reticulum Vesicles to a Replication-Permissive Vacuole. Cell. Microbiol. 2008, 10, 2416–2433. [Google Scholar] [CrossRef]
- Hay, J.C.; Chao, D.S.; Kuo, C.S.; Scheller, R.H. Protein Interactions Regulating Vesicle Transport between the Endoplasmic Reticulum and Golgi Apparatus in Mammalian Cells. Cell 1997, 89, 149–158. [Google Scholar] [CrossRef]
- Arasaki, K.; Toomre, D.; Roy, C. The Legionella Pneumophila Effector DrrA Is Sufficient to Stimulate SNARE-Dependent Membrane Fusion. Cell Host Microbe 2012, 11, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Reiling, J.H.; Olive, A.J.; Sanyal, S.; Carette, J.E.; Brummelkamp, T.R.; Ploegh, H.L.; Starnbach, M.N.; Sabatini, D.M. A CREB3–ARF4 Signalling Pathway Mediates the Response to Golgi Stress and Susceptibility to Pathogens. Nat. Cell Biol. 2013, 15, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Jeng, E.E.; Bhadkamkar, V.; Ibe, N.U.; Gause, H.; Jiang, L.; Chan, J.; Jian, R.; Jimenez-Morales, D.; Stevenson, E.; Krogan, N.J.; et al. Systematic Identification of Host Cell Regulators of Legionella Pneumophila Pathogenesis Using a Genome-Wide CRISPR Screen. Cell Host Microbe 2019, 26, 551–563. [Google Scholar] [CrossRef]
- Price, C.T.; Al-Khodor, S.; Al-Quadan, T.; Santic, M.; Habyarimana, F.; Kalia, A.; Kwaik, Y.A. Molecular Mimicry by an F-Box Effector of Legionella Pneumophila Hijacks a Conserved Polyubiquitination Machinery within Macrophages and Protozoa. PLoS Pathog. 2009, 5, e1000704. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Doms, A.G.; Cheng, E.; Kim, B.; Evans, T.R.; Machner, M.P. Host Cell-Catalyzed S-Palmitoylation Mediates Golgi Targeting of the Legionella Ubiquitin Ligase GobX. J. Biol. Chem. 2015, 290, 25766–25781. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef]
- Dreyfus, L.A. Virulence Associated Ingestion of Legionella Pneumophila by HeLa Cells. Microb. Pathog. 1987, 3, 45–52. [Google Scholar] [CrossRef]
- Becker, J.L.; Tran, D.T.; Tabak, L.A. Members of the GalNAc-T Family of Enzymes Utilize Distinct Golgi Localization Mechanisms. Glycobiology 2018, 28, 841–848. [Google Scholar] [CrossRef]
- Devlin, J.R.; Santus, W.; Mendez, J.; Peng, W.; Yu, A.; Wang, J.; Alejandro-Navarreto, X.; Kiernan, K.; Singh, M.; Jiang, P.; et al. Salmonella Enterica Serovar Typhimurium Chitinases Modulate the Intestinal Glycome and Promote Small Intestinal Invasion. PLoS Pathog. 2022, 18, e1010167. [Google Scholar] [CrossRef]
- Park, D.; Arabyan, N.; Williams, C.C.; Song, T.; Mitra, A.; Weimer, B.C.; Maverakis, E.; Lebrilla, C.B. Salmonella Typhimurium Enzymatically Landscapes the Host Intestinal Epithelial Cell (IEC) Surface Glycome to Increase Invasion. Mol. Cell. Proteom. MCP 2016, 15, 3653. [Google Scholar] [CrossRef]
- Arabyan, N.; Park, D.; Foutouhi, S.; Weis, A.M.; Huang, B.C.; Williams, C.C.; Desai, P.; Shah, J.; Jeannotte, R.; Kong, N.; et al. Salmonella Degrades the Host Glycocalyx Leading to Altered Infection and Glycan Remodeling. Sci. Rep. 2016, 6, 29525. [Google Scholar] [CrossRef]
- Arabyan, N.; Weis, A.M.; Huang, B.C.; Weimer, B.C. Implication of Sialidases in Salmonella Infection: Genome Release of Sialidase Knockout Strains from Salmonella Enterica Serovar Typhimurium LT2. Genome Announc. 2017, 5, e00341-17. [Google Scholar] [CrossRef]
- González-Morelo, K.J.; Vega-Sagardía, M.; Garrido, D. Molecular Insights Into O-Linked Glycan Utilization by Gut Microbes. Front. Microbiol. 2020, 11, 2834. [Google Scholar] [CrossRef]
- Leyn, S.A.; Gao, F.; Yang, C.; Rodionov, D.A. N-Acetylgalactosamine Utilization Pathway and Regulon in Proteobacteria. J. Biol. Chem. 2012, 287, 28047–28056. [Google Scholar] [CrossRef]
- Eisenreich, W.; Heuner, K. The Life Stage-Specific Pathometabolism of Legionella pneumophila. FEBS Lett. 2016, 590, 3868–3886. [Google Scholar] [CrossRef]
- Harada, E.; Iida, K.-I.; Shiota, S.; Nakayama, H.; Yoshida, S.-I. Glucose Metabolism in Legionella Pneumophila: Dependence on the Entner-Doudoroff Pathway and Connection with Intracellular Bacterial Growth. J. Bacteriol. 2010, 192, 2892–2899. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Macwan, V.; Heineman, R.E.-S.; Terebiznik, M.R.; Harrison, R.E. Legionella pneumophila Infection of Human Macrophages Retains Golgi Structure but Reduces O-Glycans. Pathogens 2022, 11, 908. https://doi.org/10.3390/pathogens11080908
Fu Y, Macwan V, Heineman RE-S, Terebiznik MR, Harrison RE. Legionella pneumophila Infection of Human Macrophages Retains Golgi Structure but Reduces O-Glycans. Pathogens. 2022; 11(8):908. https://doi.org/10.3390/pathogens11080908
Chicago/Turabian StyleFu, Yanlin, Vinitha Macwan, Rebecca Emily-Sue Heineman, Mauricio R. Terebiznik, and Rene E. Harrison. 2022. "Legionella pneumophila Infection of Human Macrophages Retains Golgi Structure but Reduces O-Glycans" Pathogens 11, no. 8: 908. https://doi.org/10.3390/pathogens11080908