

Re-Emergence of HMPV in Gwangju, South Korea, after the COVID-19 Pandemic

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Surveillance and Sample Collection
2.2. RNA Extraction and Real-Time PCR
2.3. Spcemens and Samples Collection
2.4. Whole Genome Sequencing
2.5. Phylogenetic Analyses
3. Results
3.1. Epidemiology of HMPV
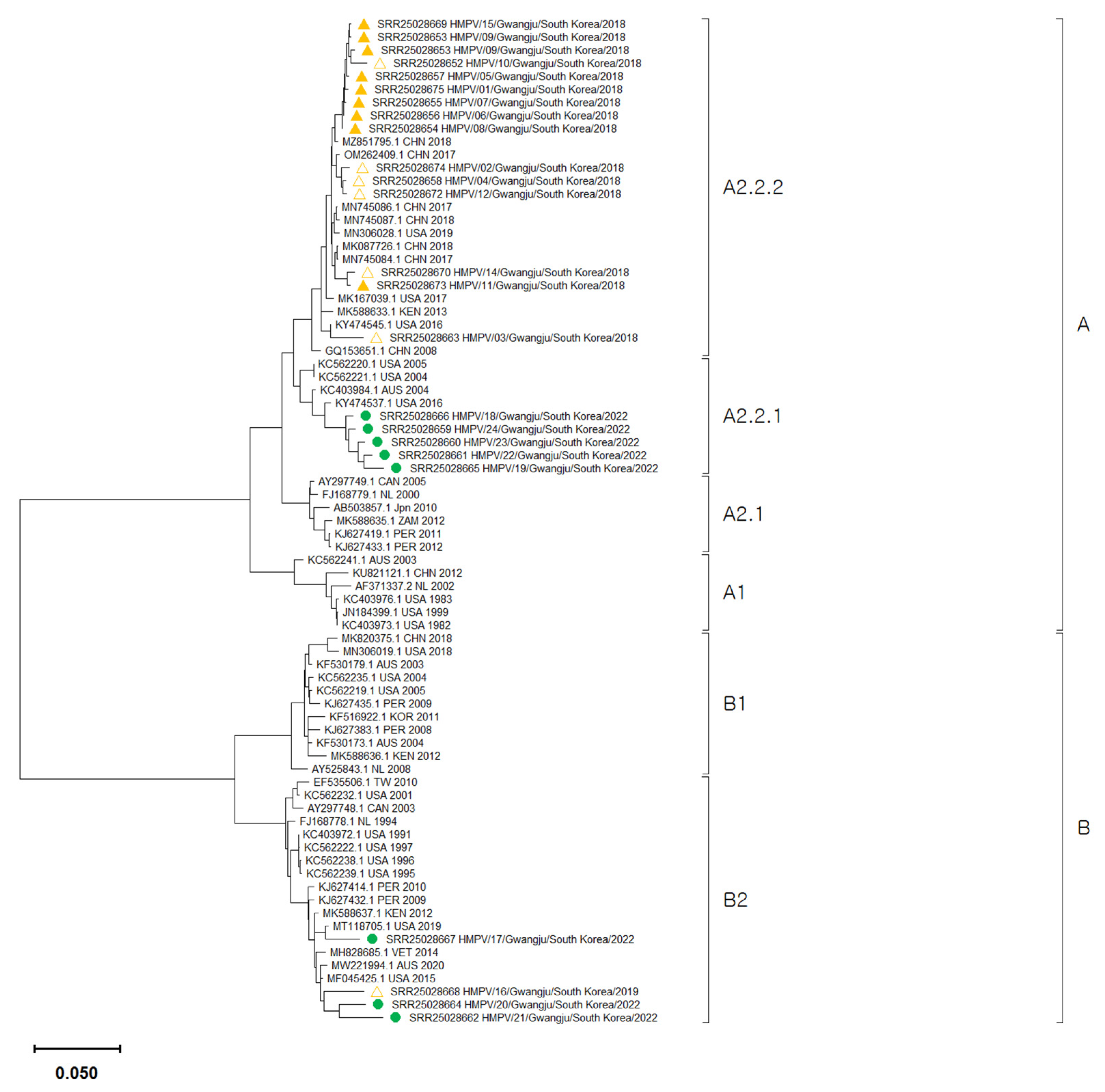
3.2. Phylogenetic Analysis of HMPV Whole Genome Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Endo, R.; Kikuta, H.; Ishiguro, N.; Ishiko, H.; Hara, M.; Takahashi, Y.; Kobayashi, K. Human metapneumovirus infection in Japanese children. J. Clin. Microbiol. 2004, 42, 126–132. [Google Scholar] [CrossRef]
- Lu, G.; Gonzalez, R.; Guo, L.; Wu, C.; Wu, J.; Vernet, G.; Paranhos-Baccalà, G.; Wang, J.; Hung, T. Large-scale seroprevalence analysis of human metapneumovirus and human respiratory syncytial virus infections in Beijing, China. Virol. J. 2011, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.E.; Williams, J.V. Human Metapneumovirus. Microbiol. Spectr. 2014, 2, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Liu, D.; Chen, D.; Tan, W.; Qiu, S.; Xu, D.; Li, X.; Liu, T.; Zhou, R. Epidemiological and clinical features of human metapneumovirus in hospitalised paediatric patients with acute respiratory illness: A cross-sectional study in Southern China, from 2013 to 2016. BMJ Open 2018, 8, e019308. [Google Scholar] [CrossRef]
- Panda, S.; Mohakud, N.K.; Pena, L.; Kumar, S. Human metapneumovirus: Review of an important respiratory pathogen. Int. J. Infect. Dis. 2014, 25, 45–52. [Google Scholar] [CrossRef]
- Van den Hoogen, B.G.; van Doornum, G.J.; Fockens, J.C.; Cornelissen, J.J.; Beyer, W.E.; Groot, R.d.; Osterhaus, A.D.; Fouchier, R.A. Prevalence and clinical symptoms of human metapneumovirus infection in hospitalized patients. J. Infect. Dis. 2003, 188, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Erdman, D.; Anderson, L.J.; Walsh, E.E. Human metapneumovirus infections in young and elderly adults. J. Infect. Dis. 2003, 187, 785–790. [Google Scholar] [CrossRef]
- Foley, D.A.; Sikazwe, C.T.; Minney-Smith, C.A.; Ernst, T.; Moore, H.C.; Nicol, M.P.; Smith, D.W.; Levy, A.; Blyth, C.C. An Unusual Resurgence of Human Metapneumovirus in Western Australia following the Reduction of Non-Pharmaceutical Interventions to Prevent SARS-CoV-2 Transmission. Viruses 2022, 14, 2135. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, B.G.; Herfst, S.; Sprong, L.; Cane, P.A.; Forleo-Neto, E.; de Swart, R.L.; Osterhaus, A.D.; Fouchier, R.A. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 2004, 10, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Nao, N.; Saikusa, M.; Sato, K.; Sekizuka, T.; Usuku, S.; Tanaka, N.; Nishimura, H.; Takeda, M. Recent Molecular Evolution of Human Metapneumovirus (HMPV): Subdivision of HMPV A2b Strains. Microorganisms 2020, 8, 1280. [Google Scholar] [CrossRef] [PubMed]
- Huck, B.; Scharf, G.; Neumann-Haefelin, D.; Puppe, W.; Weigl, J.; Falcone, V. Novel human metapneumovirus sublineage. Emerg. Infect. Dis. 2006, 12, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Saikusa, M.; Nao, N.; Kawakami, C.; Usuku, S.; Sasao, T.; Toyozawa, T.; Takeda, M.; Okubo, I. A novel 111-nucleotide duplication in the G gene of human metapneumovirus. Microbiol. Immunol. 2017, 61, 507–512. [Google Scholar] [CrossRef]
- Saikusa, M.; Kawakami, C.; Nao, N.; Takeda, M.; Usuku, S.; Sasao, T.; Nishimoto, K.; Toyozawa, T. 180-nucleotide duplication in the G gene of human metapneumovirus A2b subgroup strains circulating in Yokohama City, Japan, since 2014. Front. Microbiol. 2017, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Pinana, M.; Vila, J.; Gimferrer, L.; Valls, M.; Andres, C.; Codina, M.G.; Ramon, J.; Martin, M.C.; Fuentes, F.; Saiz, R.; et al. Novel human metapneumovirus with a 180-nucleotide duplication in the G gene. Futur. Microbiol. 2017, 12, 565–571. [Google Scholar] [CrossRef]
- Xie, Z.; Xu, J.; Ren, Y.; Cui, A.; Wang, H.; Song, J.; Zhang, Q.; Hu, M.; Xu, W.; Zhang, Y. emerging Human metapneumovirus gene duplication variants in patients with severe acute respiratory infection, China, 2017–2019. Emerg. Infect. Dis. 2021, 27, 275. [Google Scholar] [CrossRef]
- Jagusic, M.; Slovic, A.; Ivancic-Jelecki, J.; Ljubin-Sternak, S.; Vilibić-Čavlek, T.; Tabain, I.; Forcic, D. Molecular epidemiology of human respiratory syncytial virus and human metapneumovirus in hospitalized children with acute respiratory infections in Croatia, 2014–2017. Infect. Genet. Evol. 2019, 76, 104039. [Google Scholar] [CrossRef]
- Saikusa, M.; Nao, N.; Kawakami, C.; Usuku, S.; Tanaka, N.; Tahara, M.; Takeda, M.; Okubo, I. Predominant detection of the subgroup a2b human metapneumovirus strain with a 111-nucleotide duplication in the g gene in Yokohama city, Japan in 2018. Jpn. J. Infect. Dis. 2019, 72, 350–352. [Google Scholar] [CrossRef]
- Pinana, M.; Vila, J.; Maldonado, C.; Galano-Frutos, J.J.; Valls, M.; Sancho, J.; Nuvials, F.X.; Andrés, C.; Martín-Gómez, M.T.; Esperalba, J. Insights into immune evasion of human metapneumovirus: Novel 180-and 111-nucleotide duplications within viral G gene throughout 2014–2017 seasons in Barcelona, Spain. J. Clin. Virol. 2020, 132, 104590. [Google Scholar] [CrossRef]
- Rahman, M.Z.; Sumiya, M.; Sahabuddin, M.; Pell, L.G.; Gubbay, J.B.; Rahman, R.; Momtaz, F.; Azmuda, N.; Shanta, S.S.; Jahan, I. Genetic characterization of human metapneumovirus identified through community and facility-based surveillance of infants in Dhaka, Bangladesh. J. Med. Virol. 2019, 91, 549–554. [Google Scholar] [CrossRef]
- Jallow, M.M.; Fall, A.; Kiori, D.; Sy, S.; Goudiaby, D.; Barry, M.A.; Fall, M.; Niang, M.N.; Dia, N. Epidemiological, clinical and genotypic features of human Metapneumovirus in patients with influenza-like illness in Senegal, 2012 to 2016. BMC Infect. Dis. 2019, 19, 457. [Google Scholar] [CrossRef]
- Jagušić, M.; Slović, A.; Ljubin-Sternak, S.; Mlinarić-Galinović, G.; Forčić, D. Genetic diversity of human metapneumovirus in hospitalized children with acute respiratory infections in Croatia. J. Med. Virol. 2017, 89, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Neemuchwala, A.; Duvvuri, V.R.; Marchand-Austin, A.; Li, A.; Gubbay, J.B. Human metapneumovirus prevalence and molecular epidemiology in respiratory outbreaks in Ontario, Canada. J. Med. Virol. 2015, 87, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Groen, K.; van Nieuwkoop, S.; Meijer, A.; van der Veer, B.; van Kampen, J.J.; Fraaij, P.L.; Fouchier, R.A.; van den Hoogen, B.G. Emergence and Potential Extinction of Genetic Lineages of Human Metapneumovirus between 2005 and 2021. Mbio 2022, 14, e0228022. [Google Scholar] [CrossRef] [PubMed]
- Reiche, J.; Jacobsen, S.; Neubauer, K.; Hafemann, S.; Nitsche, A.; Milde, J.; Wolff, T.; Schweiger, B. Human metapneumovirus: Insights from a ten-year molecular and epidemiological analysis in Germany. PLoS ONE 2014, 9, e88342. [Google Scholar] [CrossRef]
- Oketch, J.W.; Kamau, E.; Otieno, G.P.; Otieno, J.R.; Agoti, C.N.; Nokes, D.J. Human metapneumovirus prevalence and patterns of subgroup persistence identified through surveillance of pediatric pneumonia hospital admissions in coastal Kenya, 2007–2016. BMC Infect. Dis. 2019, 19, 757. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Park, S.; Lee, I.; Park, K.S.; Kwak, E.J.; Moon, K.M.; Lee, C.K.; Bae, J.-Y.; Park, M.-S.; Song, K.-J. Genome-wide analysis of human metapneumovirus evolution. PLoS ONE 2016, 11, e0152962. [Google Scholar] [CrossRef] [PubMed]
- Cong, S.; Wang, C.; Wei, T.; Xie, Z.; Huang, Y.; Tan, J.; Chen, A.; Ma, F.; Zheng, L. Human metapneumovirus in hospitalized children with acute respiratory tract infections in Beijing, China. Infect. Genet. Evol. 2022, 106, 105386. [Google Scholar] [CrossRef]
- Agca, H.; Akalin, H.; Saglik, I.; Hacimustafaoglu, M.; Celebi, S.; Ener, B. Changing epidemiology of influenza and other respiratory viruses in the first year of COVID-19 pandemic. J. Infect. Public Health 2021, 14, 1186–1190. [Google Scholar] [CrossRef]
- Olsen, S.J.; Winn, A.K.; Budd, A.P.; Prill, M.M.; Steel, J.; Midgley, C.M.; Kniss, K.; Burns, E.; Rowe, T.; Foust, A.; et al. Changes in Influenza and Other Respiratory Virus Activity during the COVID-19 Pandemic—United States, 2020–2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1013–1019. [Google Scholar] [CrossRef]
- Lee, H.; Kim, S.H.; Cho, S.J.; Lee, Y.U.; Lee, K.; Lee, Y.P.; Seo, J.; Chung, Y.S. Genetic Analysis of HPIV3 That Emerged during the SARS-CoV-2 Pandemic in Gwangju, South Korea. Viruses 2022, 14, 1446. [Google Scholar] [CrossRef]
- Stein, M.; Cohen, H.; Nemet, I.; Atari, N.; Kliker, L.; Fratty, I.S.; Bucris, E.; Geva, M.; Mendelson, E.; Zuckerman, N. Human metapneumovirus prevalence during 2019–2021 in Israel is influenced by the COVID-19 pandemic. Int. J. Infect. Dis. 2022, 120, 205–209. [Google Scholar] [CrossRef]
- Kivit, C.; Groen, K.; Jongbloed, M.; Linssen, C.; van Loo, A.; van Gorp, E.; van Nieuwkoop, S.; van den Hoogen, B.; de Kruif, M. An off-season outbreak of human metapneumovirus infections after ending of a COVID-19 lockdown. J. Infect. 2022, 84, 722–746. [Google Scholar] [CrossRef]
- García-García, M.L.; Pérez-Arenas, E.; Pérez-Hernandez, P.; Falces-Romero, I.; Ruiz, S.; Pozo, F.; Casas, I.; Calvo, C. Human Metapneumovirus Infections during COVID-19 Pandemic, Spain. Emerg. Infect. Dis. 2023, 29, 850. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, R.L.; Kok, J.; Carter, I.; Dwyer, D.E.; Eden, J.-S. An amplicon-based approach for the whole-genome sequencing of human metapneumovirus. Viruses 2021, 13, 499. [Google Scholar] [CrossRef] [PubMed]
- Casalegno, J.S.; Ploin, D.; Cantais, A.; Masson, E.; Bard, E.; Valette, M.; Fanget, R.; Targe, S.C.; Myar-Dury, A.F.; Doret-Dion, M.; et al. Characteristics of the delayed respiratory syncytial virus epidemic, 2020/2021, Rhône Loire, France. Eurosurveillance 2021, 26, 2100630. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Wu, T.H.; Fang, Y.P.; Chang, J.C.; Wang, H.C.; Lin, S.J.; Mai, C.H.; Chang, Y.C.; Chou, T.Y. Delayed respiratory syncytial virus outbreak in 2020 in Taiwan was correlated with two novel RSV-A genotype ON1 variants. Influ. Other Respir. Viruses 2022, 16, 511–520. [Google Scholar] [CrossRef]
- Houlihan, C.F.; Frampton, D.; Ferns, R.B.; Raffle, J.; Grant, P.; Reidy, M.; Hail, L.; Thomson, K.; Mattes, F.; Kozlakidis, Z. Use of whole-genome sequencing in the investigation of a nosocomial influenza virus outbreak. J. Infect. Dis. 2018, 218, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Gymoese, P.; Sorensen, G.; Litrup, E.; Olsen, J.E.; Nielsen, E.M.; Torpdahl, M. Investigation of Outbreaks of Salmonella enterica Serovar Typhimurium and Its Monophasic Variants Using Whole-Genome Sequencing, Denmark. Emerg. Infect. Dis. 2017, 23, 1631–1639. [Google Scholar] [CrossRef]
- Piñana, M.; Vila, J.; Gimferrer, L.; Valls, M.; Andrés, C.; Ramón, J.; Codina, M.G.; del Carmen Martín, M.; Fuentes, F.; Saiz, R. Genetic variability of human metapneumovirus A strain circulating in Catalonia during the 2014–2015 and 2015–2016 seasons: A 180-nucleotide G gene duplication reported. J. Clin. Virol. 2016, 82, S125. [Google Scholar] [CrossRef]
- Wang, C.; Wei, T.; Ma, F.; Wang, H.; Guo, J.; Chen, A.; Huang, Y.; Xie, Z.; Zheng, L. Epidemiology and genotypic diversity of human metapneumovirus in paediatric patients with acute respiratory infection in Beijing, China. Virol. J. 2021, 18, 40. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Variable | 2018–2020 (n = 195) | 2022 (n = 232) | p-Value 1 | ||||
|---|---|---|---|---|---|---|---|
| Number of Patients | Number of HMPV-Positive | Prevalence of HMPV(%) | Number of Patients | Number of HMPV-Positive | Prevalence of HMPV(%) | ||
| 4264 | 195 | 4.6 | 2070 | 232 | 11.2 | <0.01 * | |
| Sex | 0.389 | ||||||
| Male | 1939 | 86 | 4.4 | 985 | 112 | 11.4 | |
| Female | 2325 | 109 | 4.7 | 1085 | 120 | 11.1 | |
| Age | |||||||
| 0–2 years | 690 | 33 | 4.8 | 666 | 65 | 9.8 | <0.01 * |
| 3–5 years | 954 | 77 | 8.1 | 481 | 96 | 20.0 | <0.01 * |
| 6–10 years | 705 | 32 | 4.5 | 204 | 43 | 21.1 | <0.01 * |
| 11–20 years | 444 | 13 | 2.9 | 209 | 10 | 4.8 | 0.164 |
| 21–40 years | 463 | 6 | 1.3 | 194 | 9 | 4.6 | 0.012 |
| 41–60 years | 451 | 13 | 2.9 | 145 | 6 | 4.1 | 0.305 |
| 60–90 years | 557 | 21 | 3.8 | 171 | 3 | 1.8 | 0.947 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.-J.; Kim, S.-H.; Lee, H.; Lee, Y.-U.; Mun, J.; Park, S.; Park, J.; Park, J.-S.; Lee, K.; Lee, C.-m.; et al. Re-Emergence of HMPV in Gwangju, South Korea, after the COVID-19 Pandemic. Pathogens 2023, 12, 1218. https://doi.org/10.3390/pathogens12101218
Cho S-J, Kim S-H, Lee H, Lee Y-U, Mun J, Park S, Park J, Park J-S, Lee K, Lee C-m, et al. Re-Emergence of HMPV in Gwangju, South Korea, after the COVID-19 Pandemic. Pathogens. 2023; 12(10):1218. https://doi.org/10.3390/pathogens12101218
Chicago/Turabian StyleCho, Sun-Ju, Sun-Hee Kim, Hongsu Lee, Yeong-Un Lee, Jeongeun Mun, Sujung Park, Jungwook Park, Ji-Su Park, Kwangho Lee, Cheong-mi Lee, and et al. 2023. "Re-Emergence of HMPV in Gwangju, South Korea, after the COVID-19 Pandemic" Pathogens 12, no. 10: 1218. https://doi.org/10.3390/pathogens12101218
APA StyleCho, S.-J., Kim, S.-H., Lee, H., Lee, Y.-U., Mun, J., Park, S., Park, J., Park, J.-S., Lee, K., Lee, C.-m., Seo, J., Kim, Y., & Chung, Y.-S. (2023). Re-Emergence of HMPV in Gwangju, South Korea, after the COVID-19 Pandemic. Pathogens, 12(10), 1218. https://doi.org/10.3390/pathogens12101218