

A Loss of Function in LprG−Rv1410c Homologues Attenuates Growth during Biofilm Formation in Mycobacterium smegmatis

Department of Pharmacological Sciences, Stony Brook University, 100 Nicolls Road, Stony Brook, NY 11794, USA
*
Authors to whom correspondence should be addressed.
Pathogens 2023, 12(12), 1375; https://doi.org/10.3390/pathogens12121375
Submission received: 1 October 2023
/
Revised: 11 November 2023
/
Accepted: 15 November 2023
/
Published: 21 November 2023
(This article belongs to the Special Issue Biology of Mycobacterial Pathogens)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:MmpL (mycobacterial membrane protein large) proteins are integral membrane proteins that have been implicated in the biosynthesis and/or transport of mycobacterial cell wall lipids. Given the cellular location of these proteins, however, it is unclear how cell wall lipids are transported beyond the inner membrane. Moreover, given that mycobacteria grow at the poles, we also do not understand how new cell wall is added in a highly localized and presumably coordinated manner. Here, we examine the relationship between two lipid transport pathways associated with the proteins MmpL11 and LprG−Rv1410c. The lipoprotein LprG has been shown to interact with proteins involved in cell wall processes including MmpL11, which is required in biofilms for the surface localization of certain lipids. Here we report that deletion of mmpL11 (MSMEG_0241) or the lprG−rv1410c operon homologues MSMEG_3070−3069 in Mycobacterium smegmatis produced similar biofilm defects that were distinct from that of the previously reported mmpL11 transposon insertion mutant. Analysis of pellicle biofilms, bacterial growth, lipid profiles, and gene expression revealed that the biofilm phenotypes could not be directly explained by changes in the synthesis or localization of biofilm-related lipids or the expression of biofilm-related genes. Instead, the shared biofilm phenotype between ΔMSMEG_3070−3069 and ΔmmpL11 may be related to their modest growth defect, while the origins of the distinct mmpL11::Tn biofilm defect remain unclear. Our findings suggest that the mechanisms that drive pellicle biofilm formation in M. smegmatis are not connected to crosstalk between the LprG−Rv1410c and MmpL11 pathways and that any functional interaction between these proteins does not relate directly to their lipid transport function.
Keywords:
mycobacteria; cell wall; biofilms; lipid transport; MmpL11; LprG; Rv1410c; gene expression1. Introduction
Tuberculosis remains one of the top infectious killers worldwide [1]. Additionally, the continued emergence and spread of multidrug-resistant TB further demonstrates the urgent need for novel therapeutic approaches against Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB) in humans. The drug resistance of Mtb is due in part to a well-armored cell wall, which is unique in composition and architecture. The outer membrane consists of an inner leaflet of very long chain mycolic acids covalently bound to an underlying arabinogalactan-peptidoglycan layer, and an outer leaflet composed of diverse noncovalently associated lipids such as trehalose 6,6′-dimycolate (TDM), phthiocerol dimycocerosate (PDIM), and sulfolipids [2,3]. Because its structure is distinct from the outer membrane of Gram-negative bacteria, this outer layer is commonly referred to as the mycomembrane.
Mycomembrane lipids are positioned to play important roles in intercellular interactions. Indeed, disruptions in lipid biosynthesis or trafficking attenuate pathogenesis, as has been reviewed elsewhere [4,5,6]. Perturbations in mycobacterial lipids are also correlated with defects in the formation of biofilms [7,8,9,10,11,12,13]. Broadly defined, biofilms are communities of microorganisms that are associated with a self-secreted extracellular matrix that is thought to serve structural and communication roles [14]. These communities can range from surface-attached colonies to pellicles at the air–liquid interface, with the latter being one of the most studied models for mycobacterial biofilms [15]. Importantly, several mycobacterial species have been shown to form biofilms in the clinical setting. The fast-growing non-tuberculous mycobacterium (NTM) M. abscessus forms biofilms within the thickened alveolar walls and airways of cystic fibrosis patients [16], as well as in the lung cavity of a patient with chronic obstructive pulmonary disease [17]. Yamazaki et al. demonstrated a role for the biofilms of another NTM, M. avium, in bronchiolar and bronchial infections [18]. Mtb was recently found to form biofilms in human lung samples with TB, and in this state Mtb was protected from chemotherapeutic agents [19]. Moreover, mycobacterial species in this biofilm state have been found to be more resistant to antibiotics [20,21]. Overall, the relevance of biofilms to clinical infections and treatment motivates investigating the genetic and molecular mechanisms by which they form.
Diverse factors drive mycobacterial biofilm formation, as has been reviewed elsewhere [22,23,24] and in detail for multiple mycobacterial species by Chakraborty and Kumar [25]. For the two most highly studied species, M. tuberculosis and M. smegmatis, the most consistent association has been with lipids, including free mycolic acids (FMA) [11,13], mycolyl diacylglycerides (MDAG) [26], monomeromycolyl diacylglycerides (MMDAG) [7,27], mycolate wax esters (MWE) [7,8,27] and, in M. smegmatis, glycopeptidolipids (GPL) [9,28]. Genetic alterations associated with changes in lipids can affect stages that are commonly defined for bacterial biofilms and are shared by mycobacteria: substratum attachment, intercellular aggregation, and architecture maturation. For example, in M. smegmatis, lsr2 disruption is required for intracellular aggregation, whereas groEl1 is required for maturation. Further, groEL1 has an unexpected role in stabilizing FAS II enzymes KasA/KasB, whose activity feeds into mycolic acid biosynthesis; the loss of groEL1 leads to the production of short chain mycolates (thought to be precursors to FMAs) [12]. The loss of lsr2 is suppressed by mutations in the GPL biosynthesis gene mps [29]. This contradicts studies showing that GPL is required for biofilm formation [9,28], but suggests that this process depends on the integration and timing of diverse factors, and that any one factor, such as a single lipid class, cannot necessarily serve as a universal marker. Indeed, other studies have identified correlations with assorted genes and lipids: Loss of function in enzymes that make the second messengers (pp)pGpp (rel) or c-di-GMP (dcpA) are defective for biofilm formation and show reductions in total levels of not only GPLs, but also other polar lipids [30]. Notably, the involvement of lsr2 in biofilms was uncovered via a transposon insertion library screen in M. smegmatis [26]. Additional prospective studies have sought to identify other factors that influence mycobacterial biofilm formation through not only transposon insertion library screens [10,31,32] in other species, but also gene expression profiling [10,33,34], and comparative proteomics [35,36]. These have further underscored the association of lipids with the identification of additional genes and proteins related to lipid biosynthesis.
Overall, the correlations between membrane lipid composition and biofilm formation highlight lipid biosynthesis and transport as targets for disrupting these antibiotic-resistant cell aggregates. Many of the enzymes that produce mycobacterial lipids have been identified, but the mechanisms by which these products reach the cell wall and mycomembrane remain poorly understood. Of the few pathways that have been characterized, the lipoprotein LprG and the co-cistronic integral membrane transporter Rv1410c are conserved across mycobacteria and are associated with the transport of triacyl lipids such as triacylglyceride and lipoarabinomannan [37,38,39]. Intriguingly, LprG has been shown to interact in vitro with the periplasmic domains of MmpL11 [40], which is involved in biofilm maturation and the surface localization of mycolic acid-containing lipids in biofilms such as MWE and MMDAG [7,27]. This evidence for a physical interaction led us to investigate the possible functional interaction between these pathways.
In this study, our aim was to investigate the potential overlapping roles of LprG−Rv1410c and MmpL11 in lipid transport and biofilm formation. Based on previous work, we focused these studies on M. smegmatis and the effects of disrupting the homologues MSMEG_3070−3069 (lprG−rv1410c) and mmpL11 (MSMEG_0241; based on prevailing nomenclature, we use mmpL11 to designate both the M. smegmatis and Mtb homologues throughout). We found that the targeted deletion of mmpL11 or MSMEG_3070−3069 led to similar biofilm defects that were distinct from that of the previously reported mmpL11 transposon insertion mutant [7] and were not correlated with shared changes in the total or cell-surface levels of biofilm-related lipids. Instead, we found that differences in mmpL11 gene expression could underlie the distinct biofilm phenotype between ΔmmpL11 and mmpL11::Tn, and a modest growth defect could explain the biofilm defect shared between ΔMSMEG_3070−3069 and ΔmmpL11. Our findings thus suggest that although LprG and MmpL11 may physically interact in vitro [40], the biofilm defects in the ΔMSMEG_3070−3069, ΔmmpL11, and mmpL11::Tn strains are not due to overlapping functions of these loci in lipid transport.
2. Materials and Methods
2.1. Bacterial Strains, Culture Media, and Culture Conditions
Bacterial strains used in this study are listed in Table S1. Mycobacterium smegmatis mc2155 served as the parent (wild-type) strain. For planktonic growth, M. smegmatis was cultured at 37 °C with agitation at 250 rpm in Middlebrook 7H9 broth supplemented with 10% albumin/dextrose/catalase (ADC), 0.5% glycerol, and 0.05% Tween 80 (all % are v/v unless otherwise indicated). For growth on solid medium, unless otherwise indicated, M. smegmatis was plated on Middlebrook 7H11 agar containing 10% ADC, 0.5% glycerol, and 0.05% Tween 80. When required, hygromycin, kanamycin, and/or zeocin were added to the growth medium at 50, 25, or 10 µg/mL, respectively.
2.2. Construction of Mutant and Complement M. smegmatis Strains
All primers and plasmids used in the construction of mutant and complement strains of mmpL11 are listed in Table S2. The ΔMSMEG_3070−3069, ΔMSMEG_3070−3069::lprG, ΔMSMEG_3070−3069::rv1410c, ΔMSMEG_3070−3069::lprG−rv1410c and wild-type parent M. smegmatis strains were a gift from Eric Rubin [39,41]. The mmpL11::Tn, mmpL11::Tn::mmpL11Msm, mmpL11::Tn::mmpL11Mtb and wild-type parent M. smegmatis strains were a gift from Georgiana Purdy [7]. A ΔmmpL11 (ΔMSMEG_0241) strain was generated via recombineering [42] (Figure S1A). Briefly, 125 bp fragments upstream and downstream of mmpL11 were PCR-amplified from genomic M. smegmatis mc2155 DNA using primers omlp741, omlp742, omlp743, and omlp744, and the products were inserted into the pJSC407 plasmid (a gift from Jeffrey Cox) flanking a hygromycin resistance gene. The resulting plasmid pMLP082 was sequence-verified. The recombineering substrate was prepared via PCR amplification using primers omlp741 and omlp744 and gel purified. Wild-type mc2155 containing the pNIT-RecET-SacB-Kan plasmid (a gift from Christopher Sassetti) was cultured to OD600 ~0.7 and the recombinase was then induced for 3 h with 10 µM isovaleronitrile. Electrocompetent cells prepared via several washes with 10% (v/v) sterile glycerol were electroporated with 1 µg of recombineering substrate and plated on 7H11/10% ADC/hygromycin agar plates to select for recombinants. Colonies were screened for recombination using PCR primers omlp745, ojcs240, omlp741, and omlp744. A confirmed clone was cured of the pNIT-RecET-SacB-Kan plasmid by screening for hygromycin resistance and kanamycin sensitivity on 7H11/10% ADC agar plates containing the appropriate antibiotic.
For complementation, the M. smegmatis mmpL11 gene was amplified using primers omlp745 and omlp746, and cloned into pMV306 using In-Fusion cloning (Takara Bio, Mountain View, CA, USA). The resulting plasmid pMLP083 was sequence-verified and subsequently electroporated into ΔmmpL11 to generate ΔmmpL11::mmpL11Msm.
2.3. Biofilm Growth and CFU Enumeration
For biofilm assays, M. smegmatis strains were cultured as previously described [7]. Briefly, M. smegmatis was inoculated at OD600 0.05 in Sauton’s medium, without Tween 80, in polystyrene Petri dishes (100 mm × 15 mm) and incubated at 30 °C without disturbance for up to 5 days. Sauton’s medium contained 0.5 g/L monobasic potassium phosphate, 0.5 g/L anhydrous magnesium sulfate, 4.0 g/L L-asparagine, 0.05 g/L ferric ammonium citrate, 2.0 g/L anhydrous citric acid, 4.76% glycerol, and 1 mg/L zinc sulfate heptahydrate at pH 7.0. To enumerate viable bacteria grown under biofilm-inducing conditions, pellicle biofilms were harvested 2 days or 4 days after inoculation via centrifugation at 3000× g for 10 min. Pellets were resuspended in 5 mL PBS with 0.1% Tween 80, and 1 mL of 3 mm glass beads (Fisher, Boston, MA, USA) were added to mechanically disrupt the biofilms via manual agitation. The bacteria were then further dispersed by passing the cells through a tuberculin syringe (BD; REF 309626) seven times. Following serial dilution in PBS/0.1% Tween 80, 100 µL of each dilution (10−6 to 10−9) was plated on Middlebrook 7H10/10% ADC/0.05% glycerol agar plates. Colony forming units (CFU) were enumerated after 3 days and statistical analyses were performed using GraphPad Prism version 10.
2.4. Lipid Extraction and Analysis
“Harsh” surface lipid extraction was performed as previously described [27]. Briefly, pellicle biofilms were harvested 5 days after inoculation via centrifugation at 3000× g for 10 min, resuspended in 5 mL hexanes and sonicated at 55 °C for 15 min. The extracts were then clarified via centrifugation at 1000× g for 10 min and dried under nitrogen gas at 30 °C (Biotage TurboVap LV). For “gentle” surface lipid extraction, pellicle biofilms were harvested 5 days after inoculation via centrifugation at 3000× g for 10 min, resuspended in 5 mL hexanes and vortexed for 30 s. The extracts were then clarified via centrifugation at 1000× g for 10 min and the supernatants were dried under nitrogen gas at 30 °C (Biotage TurboVap LV). To extract total lipids, the same procedure as gentle hexane extraction was used, except that the harvested biofilms were resuspended in 5 mL of chloroform/methanol (2:1, v/v).
For analysis via thin-layer chromatography, surface and total lipid extracts were resuspended in chloroform/methanol (2:1, v/v) and spotted onto silica plates (MilliporeSigma, Chicago, IL, USA 1.05553.0001). Loads were normalized according to dry weight. Free mycolates (FM) were resolved with chloroform/methanol (96:4, v/v) [11]. Mycolate wax ester (MWE) and monomeromycolyl diacylglycerol (MMDAG) were resolved with toluene/acetone (99:1, v/v) [7]. Trehalose dimycolate (TDM), trehalose monomycolate (TMM), phosphatidylethanolamine (PE), phosphatidylinositol (PI), cardiolipin (CL), and phosphatidylinositolmannosides (PIMs) were resolved with chloroform/methanol/water (30:8:1 v/v) [43]. Lipids were visualized via immersion in 10% phosphomolybdic acid in ethanol and charred via heating. The amounts of FM, MWE, MMDAG, TDM, TMM, PE, PI, CL, and PIMs, as a percentage of the total signal for each sample, were calculated via densitometry using ImageJ [44], and the mean ± standard deviation (S.D.) from three independent experiments were analyzed. Statistical analysis was performed using GraphPad Prism version 10.
2.5. RNA Extraction and RT-qPCR
All oligonucleotides used for RT-qPCR are listed in Table S2. Pellicle biofilms were harvested 5 days after inoculation via centrifugation at 3000× g for 10 min and the pellets were resuspended in 1 mL TRIzol LS (Thermo Fisher Scientific, Waltham, MA, USA) and stored at −80 °C. TRIzol resuspensions were thawed on ice and subsequently lysed via bead beating (BeadRupter 12, OMNI International, Kennesaw, GA, USA) using 0.1 mm zirconia/silica beads (Biospec, Bartlesville, OK, USA) for 30 s at 6 m/s followed by 5 min incubation on ice for a total of 4 cycles. The lysates were clarified via centrifugation at 12,000× g at 4 °C for 10 min and total RNA was extracted and purified from the aqueous phase using chloroform/isoamyl alcohol (24:1, v/v) and the Qiagen Rneasy kit, respectively. Successful isolation of total RNA was confirmed via visual inspection using 2% agarose gel electrophoresis. DNA contamination was removed using the TurboDNAse free kit (Thermo Fisher Scientific, Waltham, MA, USA) and 1 µg of RNA was used for cDNA synthesis with the Verso cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. RT-qPCR was performed on a LightCycler 480 (Roche, Indianapolis, IN, USA) with SYBR Green Master mix (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. Relative expression of target genes was calculated using the Pfaffl method [45].
3. Results and Discussion
3.1. Loss of MSMEG_3070 3069 or mmpL11 Leads to Similar Defects in Biofilm Formation
We hypothesized that if LprG−Rv1410c functions parallel to or downstream of MmpL11, loss of function would lead to a similar defect in biofilm formation. To test this, we used a previously reported M. smegmatis ΔMSMEG_3070−3069 deletion strain [39,41]. To enable direct comparison, we constructed a targeted deletion of mmpL11 in the same parent wild-type strain. Following established protocols [7], we compared the development of pellicle biofilms over a total of 5 days for these and corresponding complement strains (Figure 1A and Figure S2). The wild-type strain formed an immature pellicle biofilm at the air–liquid interface after 2 days; a monolayer-like pellicle biofilm after 3 days; and a mature biofilm with reticulations after 4 days. In contrast, both ΔMSMEG_3070−3069 and ΔmmpL11 showed a delay in pellicle biofilm formation and failed to form mature reticulated biofilms by day 4 (Figure 1A) or even day 5 (Figure S2). Importantly, both the kinetics and full maturation of biofilm formation were restored via complementation (Figure 1A and Figure S2). Since an in vitro interaction was previously detected between MmpL11 and LprG, but not with Rv1410c [40], we also assayed biofilm formation in ΔMSMEG_3070−3069, complemented with either Mtb lprG or rv1410c, rather than the full operon (Figure S3). Both singly complemented strains showed an identical phenotype to the wild-type strain, suggesting that either gene is sufficient for wild-type biofilm formation but only removal of both genes leads to a biofilm phenotype that is similar to that of ΔmmpL11.
The delayed maturation biofilm phenotype prompted us to ask whether the gene deletion strains were attenuated for growth under biofilm conditions. Indeed, we found that both ΔMSMEG_3070−3069 and ΔmmpL11 strains grew more slowly, with approximately 1-log fewer colony forming units (CFU) compared to the wild-type strain 2 days after inoculation (Figure 2A,B). Although this difference was not statistically significant 4 days after inoculation, the trend and magnitude were similar (Figure 2C,D). For both mutants, growth at both timepoints was restored via complementation (Figure 2A,C). Thus, both ΔMSMEG_3070−3069 and ΔmmpL11 display a moderate growth defect that may contribute to the observed biofilm defect.
This contrasts with M. smegmatis mmpL11::Tn, which was reported to have a biofilm maturation defect, but wild-type-like growth under similar conditions [7]. Therefore, we also assayed the biofilm and growth characteristics of the transposon insertion and corresponding complement strains. For mmpL11::Tn, biofilm formation resembled that of the parent wild-type strain until day 3 (Figure 1B and Figure S2), after which it remained an easily disturbed monolayer without reticulations (Figure 1B and Figure S2), and its growth was similar to that of the wild-type at both 2 days and 4 days post inoculation (Figure 2). While this characterization is similar to previous reports [7], it is notably distinct from the phenotype of our ΔmmpL11-targeted deletion strain, suggesting that the different methods of gene disruption have different consequences for biofilm formation and growth. Notably, mmpL11 is expressed in an operon upstream of MSMEG_0240, and transposon insertion could have different effects on MSMEG_0240 expression than gene deletion, although how expression changes would affect bacterial physiology is not obvious from the available information: MSMEG_0240 is annotated as a transcription factor, but is otherwise an uncharacterized hypothetical protein.
Finally, in our hands and in contrast to previous reports [7], complementation of mmpL11::Tn with mmpL11 from M. smegmatis did not restore biofilm formation and growth trended lower compared to the wild-type (Figure 1, Figure 2 and Figure S2). In contrast, the M. tuberculosis mmpL11 complement yielded wild-type-like biofilms and growth. Differences in how strains were complemented complicate the interpretation of these conflicting results. Complementation of mmpL11::Tn was achieved using a multi-copy plasmid with a strong constitutive promoter driving expression of M. smegmatis mmpL11, but in the Mtb mmpL11 complement, mmpL11 gene expression was driven by a native promoter from a single-copy integrating plasmid. Further, we complemented ΔmmpL11 from a single-copy integrated plasmid with a native promoter driving expression of Msm mmpL11. Overall, the parallels in biofilm formation and growth between ΔMSMEG_3070−3069 and ΔmmpL11, but contrast with mmpL11::Tn, led us to examine other aspects of these strains that could underlie these similarities and differences.
3.2. MSMEG_3070−3069 Is Not Required for the Surface Localization of Mycolate Wax Esters and Monomeromycolyl Diacylglycerides
The defect in biofilm formation in the mmpL11::Tn strain has been correlated with a biofilm- and cell surface-specific loss of certain lipids: mycolate wax esters (MWE), monomeromycolyl diacylglyceride (MMDAG), and a chromatographically resolved but chemically undefined species annotated as Lipid A [7]. This phenotype led to the assignment of MmpL11 as the transporter for these lipid classes. Changes in other mycomembrane lipids, such as disruptions in TDM synthesis and enzymatic hydrolysis by trehalose dimycolate hydrolase [11], have also been associated with biofilm defects. These results suggest a model in which protein interactions involving lipid biosynthesis enzymes modulate lipid composition and thereby, biofilm formation. Based on this model, the physical interaction between LprG and MmpL11 could modulate the transport of MmpL11-associated lipids like MWE and MMDAG and thereby affect biofilm formation. We would thus predict that ΔMSMEG_3070−3069 and ΔmmpL11 have similar changes in their surface lipids compared to the wild-type, although, based on biofilm formation and growth, possibly distinct from mmpL11::Tn. To test this hypothesis, we analyzed lipids extracted from M. smegmatis biofilms 5 days post inoculation via thin layer chromatography, thus allowing for the comparison of lipid profiles with the biofilm phenotypes shown in Figure 1A and Figure S2.
Surface-selective loss of MWE, MMDAG, and lipid A in mmpL11::Tn biofilms was previously shown by extracting lipids with hexanes and sonication with heating. To facilitate direct comparison, we applied this method to ΔMSMEG_3070−3069 and the corresponding wild-type and complement strains. Surprisingly, ΔMSMEG_3070−3069 extracts showed elevated triacylglycerides (Figure 3A). This result contradicts the prevailing model from Mtb, in which LprG/Rv1410c transport TAG to the mycomembrane, but is consistent with the accumulation of select TAG isoforms in the total lipid extracts of Mtb ΔlprG−rv1410c. One possible explanation is that the extraction method is not surface-selective: sonication was performed at 50 °C [27], which, may have led to the solubilization of lipids deeper within the mycobacterial cell envelope. We thus compared this to a gentler extraction in hexanes without sonication or heating, an approach that has been previously used to characterize lipid transport in Mtb [46,47,48]. Although TAG is considered a mycomembrane lipid based on surface-selective lipid extraction with reverse micelles, hexanes extracts obtained without sonication or heating showed markedly reduced extraction of TAGs, based on the comparison between treatment conditions and using a commercial tripalmitate standard (Figure 3B). However, when applied to mmpL11::Tn, this gentler extraction of hexanes still consistently recapitulated the previously reported loss of MWE, MMDAG, and lipid A (Figure 3B). We concluded that while this method may not exhaustively extract all surface lipids, it was more selective and therefore preferred for further analyses since it was less likely to lead to a false negative lipid transport phenotype.
We therefore proceeded to analyze selective “gentle” hexanes and total chloroform-methanol extracts for all MSMEG_3070−3069- and mmpL11-associated strains (Figure 3). For all strains, no major differences were detected in the total cellular levels of MWE, MMDAG, or lipid A (Figure 3D). In contrast, hexanes extracts revealed distinctions between the strains that did not, however, correlate with biofilm phenotypes (Figure 1, Figure 3B,C and Figure S2). First, M. smegmatis mmpL11 complementation restored MWE, MMDAG, and lipid A levels in mmpL11::Tn. Second, ΔmmpL11 only partially recapitulated mmpL11::Tn: While a reduction in MWE was evident, changes in MMDAG or lipid A were not, and this phenotype was restored via complementation with Msm mmpL11. (Because MMDAG is not well resolved in the replicate shown in Figure 3B, a replicate with a subset of strains more clearly illustrating the reduction of MMDAG for ΔmmpL11 is provided in Figure 3C.) Finally, the loss of MSMEG_3070 3069 function did not strongly affect any of the three lipid classes. These results suggest that changes in the surface localization of MWE, MMDAG, and lipid A can broadly be correlated with the loss of mmpL11 function, but not the loss of MSMEG_3070−3069 function. Consequently, the shared defect in biofilm formation cannot be explained by the changes in the surface localization of these three classes of lipids in these strains.
As noted in the introduction, free mycolic acids (FMA) have been implicated in biofilm formation through changes in the mycolic acid synthesis (specifically, the modulation of KasA activity by interaction with the chaperone GroEL1 [12]) and incorporation into the extracellular matrix (through the activity of a trehalose dimycolate hydrolase [11]). To test whether changes in FMA underlie the shared biofilm phenotype, we also analyzed free mycolic acid and trehalose (di)mycolate levels in all MSMEG_3070−3069 and mmpL11-related strains. However, no major changes in any of these lipid classes were detected (Figure 4). In summary we found no shared differences in surface nor total levels of lipids previously associated with biofilm formation that could explain the biofilm phenotypes we observed upon loss of MSMEG_3070−3069 and mmpL11.
3.3. Loss of MSMEG_3070−3069 or mmpL11 Does Not Correlate with Changes in Expression of Genes Associated with Biofilm Formation
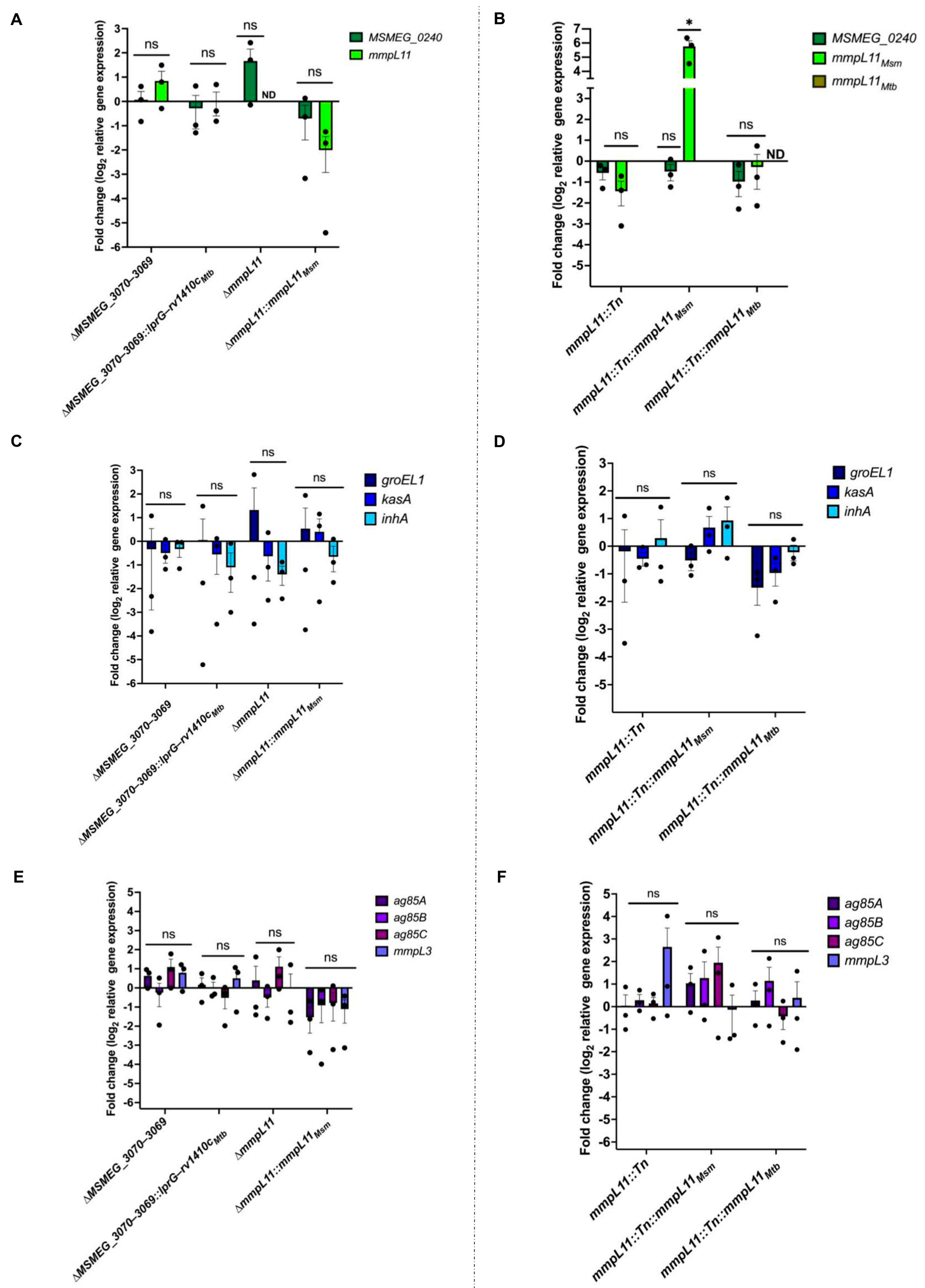
Since we found no shared differences in lipids previously associated with biofilm formation, we hypothesized that the biofilm defect in the ΔMSMEG_3070−3069, ΔmmpL11, and mmpL11::Tn strains could be due to changes in the expression of biofilm-related genes. To test this hypothesis, we measured the expression of mmpL11, the co-cistronic MSMEG_0240, and genes associated with biofilm maturation (groEl1, kasA) [12] or required for TDM synthesis (mmpL3, ag85A, ag85B, ag85C) [11]. Compared to the wild-type, no significant changes in gene expression were detected for any of these genes (Figure 5).
The only exception was mmpL11. As expected, expression was not detected in the targeted gene deletion ΔmmpL11 (Figure 5A). Low levels were found in mmpL11::Tn, as assessed using a primer pair upstream of the mapped transposon insertion site, suggesting that the insertion suppressed but did not prevent transcription (Figure 5B). However, in mmpL11::Tn complemented with M. smegmatis mmpL11, the complement was highly expressed (~64-fold increase compared to the wild-type, an average of three independent experiments) (Figure 5B). This suggests that M. smegmatis mmpL11 overexpression underlies the lack of biofilm complementation (Figure 1 and Figure S2) and the unexpected growth defect (Figure 2) in this strain. In contrast, Mtb mmpL11 expression was not detected even though this strain showed complementation of the mmpL11::Tn biofilm defect (Figure 1, Figure 5B and Figure S2). It is possible that this message is unstable when isolated, or that even very low expression is sufficient for complementation. In summary, differences in mmpL11 gene expression may contribute to the distinct biofilm phenotypes seen between ΔmmpL11 and mmpL11::Tn, but cannot explain the biofilm defect shared between ΔmmpL11 and ΔMSMEG_3070−3069.
4. Conclusions
Taken together, our findings suggest that the loss of function of MSMEG_3070−3069 and mmpL11 leads to a defect in biofilm formation that is distinct from that of a previously characterized mmpL11 transposon insertion mutant in M. smegmatis. This work also revealed that neither the difference in the biofilm phenotype between ΔmmpL11 and mmpL11::Tn, nor the similar biofilm defect in ΔMSMEG_3070−3069 and ΔmmpL11, can be directly explained by changes in the total or cell-surface levels of lipid classes that have been correlated with biofilm formation, or by changes in the expression of genes associated with biofilm formation or the synthesis of biofilm-related lipids. Instead, both null strains show a moderate growth defect that may underlie the observed delay in biofilm formation. Given that the common growth defect is not correlated with shared changes in the lipid profile, it is possible that the disruption of lipid transport in these strains dysregulates lipid metabolism in ways that similarly compromise growth. This model has been proposed for lprG−rv1410c in Mtb [39], but remains to be experimentally explored. Overall, we found that while LprG and MmpL11 may physically interact [40], these proteins do not function in overlapping lipid transport pathways that correlate with biofilm formation.
Finally, the growth and the biofilm defects in ΔmmpL11 were not shared by the mmpL11::Tn transposon mutant, but the two strains were otherwise similar in all other assays. The only clear difference was that the chemical undefined species lipid A was retained in hexanes extracts of ΔmmpL11 (Figure 3B). While we showed that this change was not correlated with differences in expression of the downstream MSMEG_0240, mutations in unlinked genes elsewhere in the chromosome or strain-specific responses to genetic modification could be responsible. An alternative, but less likely, explanation is that the low-level transcription of mmpL11 in mmpL11::Tn permits the expression of an N-terminal fragment that complements growth without restoring lipid phenotypes. Nevertheless, comparison of the two strains confirms that mmpL11 is required in biofilms for the surface localization of MMDAG and MWE. Our data also provide a note of caution concerning complementation, as the multi-copy expression of mmpL11 from a constitutive promoter led to significant overexpression and could underlie the unexpected phenotypes seen when compared to the parent strain.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/pathogens12121375/s1, Figure S1: schematic of plasmids used in the study and recombineering strategy used to generate ΔmmpL11 strain; Figure S2: 5-day time course of pellicle biofilm formation at the air–liquid interface; Figure S3: 5-day time course of MSMEG_3070−3069 mutant and complement strains’ pellicle biofilm formation at the air–liquid interface; Table S1: strains used in this study; Table S2: plasmids and primers used in this study.
Author Contributions
Conceptualization, L.-M.N. and J.C.S.; resources, M.L.P.; validation, L.-M.N., M.L.P. and J.C.S.; methodology, L.-M.N., M.L.P. and J.C.S.; formal analysis, L.-M.N. and J.C.S.; investigation, L.-M.N. and J.C.S.; writing—original draft preparation, L.-M.N., M.L.P. and J.C.S.; writing—review and editing, L.-M.N. and J.C.S.; visualization, L.-M.N. and J.C.S.; supervision, J.C.S.; project administration, J.C.S.; funding acquisition, J.C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funding from the National Institutes of Health (R01 AI141513 to J.C.S. and IRACDA NY-CAPS K12GM102778 to L.-M.N.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and supplementary materials.
Acknowledgments
We thank Eric Rubin for providing the wild-type, ΔMSMEG_3070−3069, ΔMSMEG_3070−3069::lprGMtb, ΔMSMEG_3070−3069::rv1410cMtb, and ΔMSMEG_3070−3069::lprG−rv1410cMtb M. smegmatis strains. We thank Georgiana Purdy for providing the wild-type, mmpL11::Tn, mmpL11::Tn::mmpL11Msm, and mmpL11::Tn::mmpL11Mtb M. smegmatis strains and her technical assistance. We thank Jeffrey Cox for providing the pJSC407 plasmid. We thank Christopher Sassetti for providing the pNIT-RecET-SacB-Kan plasmid. We thank members of the Seeliger lab, especially Neetika Jaisinghani, for their technical assistance and helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- WHO. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Brennan, P.J.; Nikaido, H. The Envelope of Mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef]
- Hoffmann, C.; Leis, A.; Niederweis, M.; Plitzko, J.M.; Engelhardt, H. Disclosure of the Mycobacterial Outer Membrane: Cryo-Electron Tomography and Vitreous Sections Reveal the Lipid Bilayer Structure. Proc. Natl. Acad. Sci. USA 2008, 105, 3963–3967. [Google Scholar] [CrossRef]
- Jackson, M. The Mycobacterial Cell Envelope-Lipids. Cold Spring Harb. Perspect. Med. 2014, 4, a021105. [Google Scholar] [CrossRef]
- Astarie-Dequeker, C.; Nigou, J.; Passemar, C.; Guilhot, C. The Role of Mycobacterial Lipids in Host Pathogenesis. Drug Discov. Today Dis. Mech. 2010, 7, e33–e41. [Google Scholar] [CrossRef]
- Singh, P.; Rameshwaram, N.R.; Ghosh, S.; Mukhopadhyay, S. Cell Envelope Lipids in the Pathophysiology of Mycobacterium Tuberculosis. Future Microbiol. 2018, 13, 689–710. [Google Scholar] [CrossRef]
- Pacheco, S.A.; Hsu, F.-F.; Powers, K.M.; Purdy, G.E. MmpL11 Protein Transports Mycolic Acid-Containing Lipids to the Mycobacterial Cell Wall and Contributes to Biofilm Formation in Mycobacterium Smegmatis. J. Biol. Chem. 2013, 288, 24213–24222. [Google Scholar] [CrossRef]
- Wright, C.C.; Hsu, F.F.; Arnett, E.; Dunaj, J.L.; Davidson, P.M.; Pacheco, S.A.; Harriff, M.J.; Lewinsohn, D.M.; Schlesinger, L.S.; Purdy, G.E. The Mycobacterium Tuberculosis MmpL11 Cell Wall Lipid Transporter Is Important for Biofilm Formation, Intracellular Growth, and Nonreplicating Persistence. Infect. Immun. 2017, 85, e00131-17. [Google Scholar] [CrossRef]
- Recht, J.; Martínez, A.; Torello, S.; Kolter, R. Genetic Analysis of Sliding Motility in Mycobacterium Smegmatis. J. Bacteriol. 2000, 182, 4348–4351. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Danelishvili, L.; Wu, M.; Macnab, M.; Bermudez, L.E. Mycobacterium Avium Genes Associated with the Ability to Form a Biofilm. Appl. Environ. Microbiol. 2006, 72, 819–825. [Google Scholar] [CrossRef]
- Ojha, A.K.; Trivelli, X.; Guerardel, Y.; Kremer, L.; Hatfull, G.F. Enzymatic Hydrolysis of Trehalose Dimycolate Releases Free Mycolic Acids during Mycobacterial Growth in Biofilms. J. Biol. Chem. 2010, 285, 17380–17389. [Google Scholar] [CrossRef]
- Ojha, A.; Anand, M.; Bhatt, A.; Kremer, L.; Jacobs, W.R.J.; Hatfull, G.F. GroEL1: A Dedicated Chaperone Involved in Mycolic Acid Biosynthesis during Biofilm Formation in Mycobacteria. Cell 2005, 123, 861–873. [Google Scholar] [CrossRef]
- Ojha, A.K.; Baughn, A.D.; Sambandan, D.; Hsu, T.; Trivelli, X.; Guerardel, Y.; Alahari, A.; Kremer, L.; Jacobs, W.R.J.; Hatfull, G.F. Growth of Mycobacterium Tuberculosis Biofilms Containing Free Mycolic Acids and Harbouring Drug-Tolerant Bacteria. Mol. Microbiol. 2008, 69, 164–174. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The Biofilm Matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm Formation as Microbial Development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef]
- Qvist, T.; Eickhardt, S.; Kragh, K.N.; Andersen, C.B.; Iversen, M.; Høiby, N.; Bjarnsholt, T. Chronic Pulmonary Disease with Mycobacterium Abscessus Complex Is a Biofilm Infection. Eur. Respir. J. 2015, 46, 1823–1826. [Google Scholar] [CrossRef]
- Fennelly, K.P.; Ojano-Dirain, C.; Yang, Q.; Liu, L.; Lu, L.; Progulske-Fox, A.; Wang, G.P.; Antonelli, P.; Schultz, G. Biofilm Formation by Mycobacterium Abscessus in a Lung Cavity. Am. J. Respir. Crit. Care Med. 2016, 193, 692–693. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Danelishvili, L.; Wu, M.; Hidaka, E.; Katsuyama, T.; Stang, B.; Petrofsky, M.; Bildfell, R.; Bermudez, L.E. The Ability to Form Biofilm Influences Mycobacterium Avium Invasion and Translocation of Bronchial Epithelial Cells. Cell. Microbiol. 2006, 8, 806–814. [Google Scholar] [CrossRef]
- Chakraborty, P.; Bajeli, S.; Kaushal, D.; Radotra, B.D.; Kumar, A. Biofilm Formation in the Lung Contributes to Virulence and Drug Tolerance of Mycobacterium Tuberculosis. Nat. Commun. 2021, 12, 1606. [Google Scholar] [CrossRef]
- Greendyke, R.; Byrd, T.F. Differential Antibiotic Susceptibility of Mycobacterium Abscessus Variants in Biofilms and Macrophages Compared to That of Planktonic Bacteria. Antimicrob. Agents Chemother. 2008, 52, 2019–2026. [Google Scholar] [CrossRef]
- Bardouniotis, E.; Ceri, H.; Olson, M.E. Biofilm Formation and Biocide Susceptibility Testing of Mycobacterium Fortuitum and Mycobacterium Marinum. Curr. Microbiol. 2003, 46, 28–32. [Google Scholar] [CrossRef]
- Viljoen, A.; Dufrêne, Y.F.; Nigou, J. Mycobacterial Adhesion: From Hydrophobic to Receptor-Ligand Interactions. Microorganisms 2022, 10, 454. [Google Scholar] [CrossRef]
- Richards, J.P.; Ojha, A.K. Mycobacterial Biofilms. Microbiol. Spectr. 2014, 2, 1–11. [Google Scholar] [CrossRef]
- Esteban, J.; García-Coca, M. Mycobacterium Biofilms. Front. Microbiol. 2018, 8, 2651. [Google Scholar] [CrossRef]
- Chakraborty, P.; Kumar, A. The Extracellular Matrix of Mycobacterial Biofilms: Could We Shorten the Treatment of Mycobacterial Infections? Microb. Cell 2019, 6, 105–122. [Google Scholar] [CrossRef]
- Chen, J.M.; German, G.J.; Alexander, D.C.; Ren, H.; Tan, T.; Liu, J. Roles of Lsr2 in Colony Morphology and Biofilm Formation of Mycobacterium Smegmatis. J. Bacteriol. 2006, 188, 633–641. [Google Scholar] [CrossRef]
- Melly, G.C.; Stokas, H.; Davidson, P.M.; Roma, J.S.; Rhodes, H.L.; Purdy, G.E. Identification of Residues Important for M. Tuberculosis MmpL11 Function Reveals That Function Is Modulated by Phosphorylation in the C-Terminal Domain. Mol. Microbiol. 2021, 115, 208–221. [Google Scholar] [CrossRef]
- Recht, J.; Kolter, R. Glycopeptidolipid Acetylation Affects Sliding Motility and Biofilm Formation in Mycobacterium Smegmatis. J. Bacteriol. 2001, 183, 5718–5724. [Google Scholar] [CrossRef]
- Yang, Y.; Thomas, J.; Li, Y.; Vilchèze, C.; Derbyshire, K.M.; Jacobs, W.R.J.; Ojha, A.K. Defining a Temporal Order of Genetic Requirements for Development of Mycobacterial Biofilms. Mol. Microbiol. 2017, 105, 794–809. [Google Scholar] [CrossRef]
- Gupta, K.R.; Kasetty, S.; Chatterji, D. Novel Functions of (p)ppGpp and Cyclic Di-GMP in Mycobacterial Physiology Revealed by Phenotype Microarray Analysis of Wild-Type and Isogenic Strains of Mycobacterium Smegmatis. Appl. Environ. Microbiol. 2015, 81, 2571–2578. [Google Scholar] [CrossRef]
- Richards, J.P.; Cai, W.; Zill, N.A.; Zhang, W.; Ojha, A.K. Adaptation of Mycobacterium Tuberculosis to Biofilm Growth Is Genetically Linked to Drug Tolerance. Antimicrob. Agents Chemother. 2019, 63, e01213-19. [Google Scholar] [CrossRef]
- Pang, J.M.; Layre, E.; Sweet, L.; Sherrid, A.; Moody, D.B.; Ojha, A.; Sherman, D.R. The Polyketide Pks1 Contributes to Biofilm Formation in Mycobacterium Tuberculosis. J. Bacteriol. 2012, 194, 715–721. [Google Scholar] [CrossRef]
- Dokic, A.; Peterson, E.; Arrieta-Ortiz, M.L.; Pan, M.; Di Maio, A.; Baliga, N.; Bhatt, A. Mycobacterium Abscessus Biofilms Produce an Extracellular Matrix and Have a Distinct Mycolic Acid Profile. Cell Surf. 2021, 7, 100051. [Google Scholar] [CrossRef]
- Belardinelli, J.M.; Li, W.; Avanzi, C.; Angala, S.K.; Lian, E.; Wiersma, C.J.; Palčeková, Z.; Martin, K.H.; Angala, B.; de Moura, V.C.N.; et al. Unique Features of Mycobacterium Abscessus Biofilms Formed in Synthetic Cystic Fibrosis Medium. Front. Microbiol. 2021, 12, 743126. [Google Scholar] [CrossRef]
- Savijoki, K.; Myllymäki, H.; Luukinen, H.; Paulamäki, L.; Vanha-Aho, L.-M.; Svorjova, A.; Miettinen, I.; Fallarero, A.; Ihalainen, T.O.; Yli-Kauhaluoma, J.; et al. Surface-Shaving Proteomics of Mycobacterium Marinum Identifies Biofilm Subtype-Specific Changes Affecting Virulence, Tolerance, and Persistence. mSystems 2021, 6, e0050021. [Google Scholar] [CrossRef]
- Sharma, A.; Bansal, S.; Kumari, N.; Vashistt, J.; Shrivastava, R. Comparative Proteomic Investigation Unravels the Pathobiology of Mycobacterium Fortuitum Biofilm. Appl. Microbiol. Biotechnol. 2023, 107, 6029–6046. [Google Scholar] [CrossRef]
- Shukla, S.; Richardson, E.T.; Athman, J.J.; Shi, L.; Wearsch, P.A.; McDonald, D.; Banaei, N.; Boom, W.H.; Jackson, M.; Harding, C.V. Mycobacterium Tuberculosis Lipoprotein LprG Binds Lipoarabinomannan and Determines Its Cell Envelope Localization to Control Phagolysosomal Fusion. PLoS Pathog. 2014, 10, e1004471. [Google Scholar] [CrossRef]
- Gaur, R.L.; Ren, K.; Blumenthal, A.; Bhamidi, S.; González-Nilo, F.D.; Jackson, M.; Zare, R.N.; Ehrt, S.; Ernst, J.D.; Banaei, N. LprG-Mediated Surface Expression of Lipoarabinomannan Is Essential for Virulence of Mycobacterium Tuberculosis. PLoS Pathog. 2014, 10, e1004376. [Google Scholar] [CrossRef]
- Martinot, A.J.; Farrow, M.; Bai, L.; Layre, E.; Cheng, T.-Y.; Tsai, J.H.; Iqbal, J.; Annand, J.W.; Sullivan, Z.A.; Hussain, M.M.; et al. Mycobacterial Metabolic Syndrome: LprG and Rv1410 Regulate Triacylglyceride Levels, Growth Rate and Virulence in Mycobacterium Tuberculosis. PLoS Pathog. 2016, 12, e1005351. [Google Scholar] [CrossRef]
- Melly, G.C.; Stokas, H.; Dunaj, J.L.; Hsu, F.-F.; Rajavel, M.; Su, C.-C.; Yu, E.W.; Purdy, G.E. Structural and Functional Evidence That Lipoprotein LpqN Supports Cell Envelope Biogenesis in M. Tuberculosis. J. Biol. Chem. 2019, 294, 15711–15723. [Google Scholar] [CrossRef]
- Farrow, M.F.; Rubin, E.J. Function of a Mycobacterial Major Facilitator Superfamily Pump Requires a Membrane-Associated Lipoprotein. J. Bacteriol. 2008, 190, 1783–1791. [Google Scholar] [CrossRef]
- Murphy, K.C.; Papavinasasundaram, K.; Sassetti, C.M. Mycobacterial Recombineering. Methods Mol. Biol. 2015, 1285, 177–199. [Google Scholar] [CrossRef]
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial Outer Membrane Is a Lipid Bilayer and the Inner Membrane Is Unusually Rich in Diacyl Phosphatidylinositol Dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Touchette, M.H.; Holsclaw, C.M.; Previti, M.L.; Solomon, V.C.; Leary, J.A.; Bertozzi, C.R.; Seeliger, J.C. The Rv1184c Locus Encodes Chp2, an Acyltransferase in Mycobacterium Tuberculosis Polyacyltrehalose Lipid Biosynthesis. J. Bacteriol. 2015, 197, 201–210. [Google Scholar] [CrossRef]
- Converse, S.E.; Mougous, J.D.; Leavell, M.D.; Leary, J.A.; Bertozzi, C.R.; Cox, J.S. MmpL8 Is Required for Sulfolipid-1 Biosynthesis and Mycobacterium Tuberculosis Virulence. Proc. Natl. Acad. Sci. USA 2003, 100, 6121–6126. [Google Scholar] [CrossRef]
- Seeliger, J.C.; Holsclaw, C.M.; Schelle, M.W.; Botyanszki, Z.; Gilmore, S.A.; Tully, S.E.; Niederweis, M.; Cravatt, B.F.; Leary, J.A.; Bertozzi, C.R. Elucidation and Chemical Modulation of Sulfolipid-1 Biosynthesis in Mycobacterium Tuberculosis. J. Biol. Chem. 2012, 287, 7990–8000. [Google Scholar] [CrossRef]
Figure 1.
MSMEG_3070−3069 and mmpL11 mutants displayed impaired biofilm formation. (A,B) Pellicle biofilm formation at the air–liquid interface at 2 and 4 days after inoculation for (A) parent wild-type, ΔMSMEG_3070−3069, ΔmmpL11, and associated complement strains and (B) parent wild-type, mmpL::Tn, and associated complement strains. Equal numbers of bacteria were inoculated in Sauton’s medium, without Tween 80, in polystyrene dishes and the plates were incubated at 30 °C without disturbance for 5 days. Data shown are representative of three biological replicates.
Figure 1.
MSMEG_3070−3069 and mmpL11 mutants displayed impaired biofilm formation. (A,B) Pellicle biofilm formation at the air–liquid interface at 2 and 4 days after inoculation for (A) parent wild-type, ΔMSMEG_3070−3069, ΔmmpL11, and associated complement strains and (B) parent wild-type, mmpL::Tn, and associated complement strains. Equal numbers of bacteria were inoculated in Sauton’s medium, without Tween 80, in polystyrene dishes and the plates were incubated at 30 °C without disturbance for 5 days. Data shown are representative of three biological replicates.

Figure 2.
MSMEG_3070−3069 and ΔmmpL11 show delayed growth compared to the wild-type during biofilm formation. Growth was assessed (A,B) 2 days and (C,D) 4 days after biofilm inoculation by (A,C) spotting serial dilutions on 7H10/ADC/glycerol agar or (B,D) enumerating CFU. For (A,C), data representative of three independent experiments are shown. For (B,D), the data shown are the mean ± S.D. of three independent experiments. Statistical significance was determined using one-way ANOVA (Dunnett’s test). (**, p = 0.0021; ***, p = 0.0002; ns: not significant).
Figure 2.
MSMEG_3070−3069 and ΔmmpL11 show delayed growth compared to the wild-type during biofilm formation. Growth was assessed (A,B) 2 days and (C,D) 4 days after biofilm inoculation by (A,C) spotting serial dilutions on 7H10/ADC/glycerol agar or (B,D) enumerating CFU. For (A,C), data representative of three independent experiments are shown. For (B,D), the data shown are the mean ± S.D. of three independent experiments. Statistical significance was determined using one-way ANOVA (Dunnett’s test). (**, p = 0.0021; ***, p = 0.0002; ns: not significant).
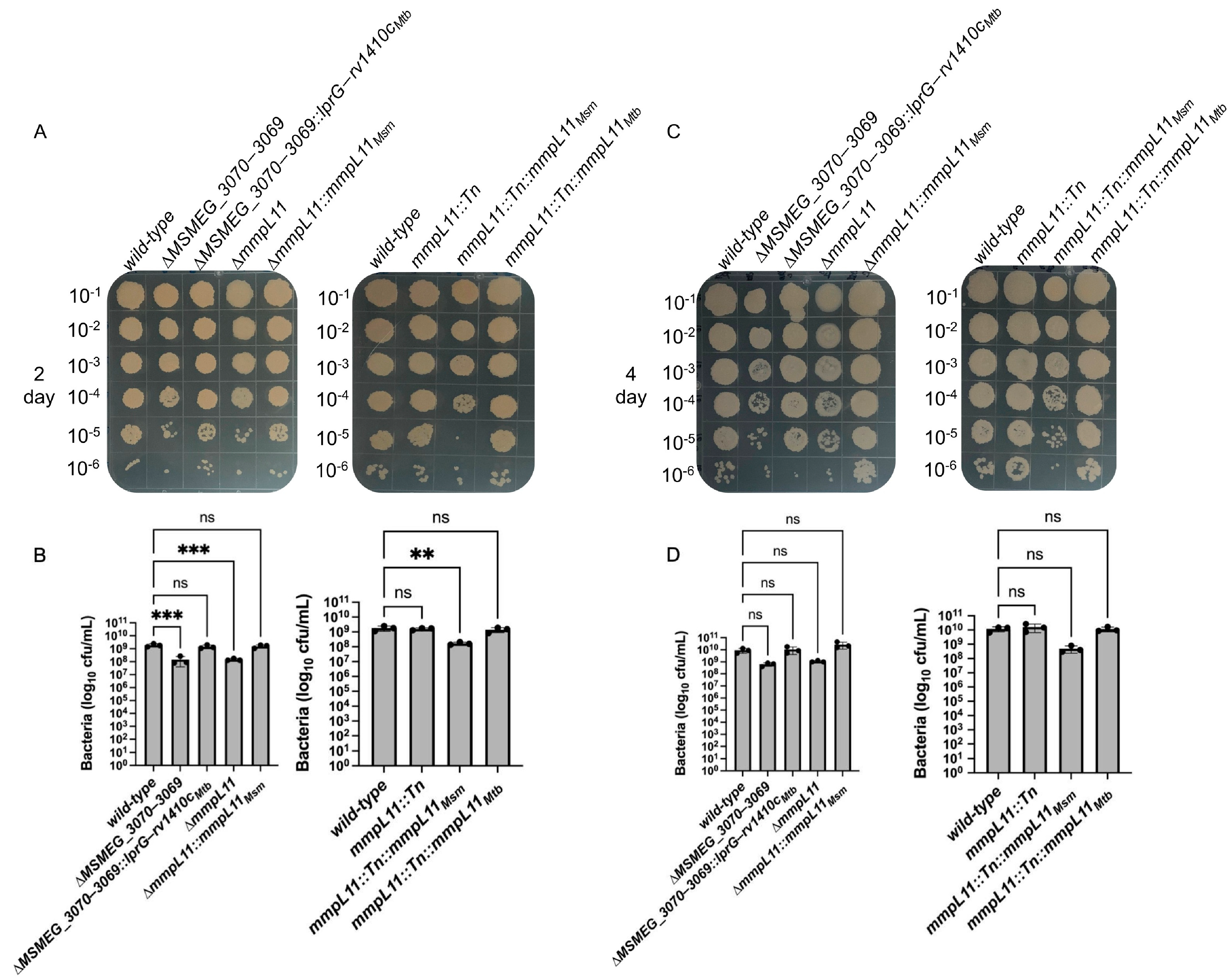
Figure 3.
MSMEG_3070−3069 mutant has an altered “harsh” hexane extract surface- exposed lipid profile of Lipid A, MWE, MMDAG and TAG but does not have an altered “gentle” hexane extract surface-exposed lipid profile. (A) 195 µg of surface lipid extracts, (B,C) 200 µg of surface lipid extracts and (D) 200 µg of total lipid extracts were resolved by TLC in toluene:acetone (99:1, v/v). Tripalmitate (labeled as TAG) was used as a migration standard. Lipid A, MWE and MMDAG were assigned by comparison to previously published TLCs performed under the same conditions [7,27]. TLC plates were immersed in 10% phosphomolybdic acid and charred to visualize lipids. The line near the top of the plates indicates the solvent front. Data shown represents one biological replicate.
Figure 3.
MSMEG_3070−3069 mutant has an altered “harsh” hexane extract surface- exposed lipid profile of Lipid A, MWE, MMDAG and TAG but does not have an altered “gentle” hexane extract surface-exposed lipid profile. (A) 195 µg of surface lipid extracts, (B,C) 200 µg of surface lipid extracts and (D) 200 µg of total lipid extracts were resolved by TLC in toluene:acetone (99:1, v/v). Tripalmitate (labeled as TAG) was used as a migration standard. Lipid A, MWE and MMDAG were assigned by comparison to previously published TLCs performed under the same conditions [7,27]. TLC plates were immersed in 10% phosphomolybdic acid and charred to visualize lipids. The line near the top of the plates indicates the solvent front. Data shown represents one biological replicate.

Figure 4.
The free mycolic acid lipid profile in the total and surface lipid extracts, and the trehalose dimycolate (TDM) and trehalose monomycolate (TMM) lipid profiles in the total lipid extracts are not altered in any strain. (A) 50 µg total lipid and (B) 50 µg surface lipid extracts were resolved via TLC in a chloroform: methanol (96:4, v/v) solvent system. (C) 50 µg total lipid extracts were resolved via TLC in chloroform: methanol: water (30:8:1, v/v). TLC plates were immersed in 10% phosphomolybdic acid and charred to visualize lipids. The line near the top of the plate indicates the solvent front. TDM: trehalose dimycolate, TMM: trehalose monomycolate, PE: phosphatidylethanolamine, CL: cardiolipin, PIMs: phosphatidylinositolmannosides. Data shown represent one replicate.
Figure 4.
The free mycolic acid lipid profile in the total and surface lipid extracts, and the trehalose dimycolate (TDM) and trehalose monomycolate (TMM) lipid profiles in the total lipid extracts are not altered in any strain. (A) 50 µg total lipid and (B) 50 µg surface lipid extracts were resolved via TLC in a chloroform: methanol (96:4, v/v) solvent system. (C) 50 µg total lipid extracts were resolved via TLC in chloroform: methanol: water (30:8:1, v/v). TLC plates were immersed in 10% phosphomolybdic acid and charred to visualize lipids. The line near the top of the plate indicates the solvent front. TDM: trehalose dimycolate, TMM: trehalose monomycolate, PE: phosphatidylethanolamine, CL: cardiolipin, PIMs: phosphatidylinositolmannosides. Data shown represent one replicate.

Figure 5.
RT-qPCR analysis of genes connected to biofilm formation in M. smegmatis. RT-qPCR analysis of (A,B) MSMEG_0240 and mmpL11, (C,D) groEL1, kasA, and inhA and (E,F) ag85A, ag85B, ag85C, and mmpL3 in the wild-type, mutant, and complement strains. Changes in gene expression are relative to the appropriate wild-type parent strain control and were calculated using the Pfaffl method [45]. The housekeeping gene sigA was used as the endogenous reference for normalization between samples. Data represent mean ± SEM of three independent biological replicates. Statistical significance was determined using one-way ANOVA (Dunnett’s test) in GraphPad Prism version 10. (*, p = 0.03; ns: not significant). ND: not detected.
Figure 5.
RT-qPCR analysis of genes connected to biofilm formation in M. smegmatis. RT-qPCR analysis of (A,B) MSMEG_0240 and mmpL11, (C,D) groEL1, kasA, and inhA and (E,F) ag85A, ag85B, ag85C, and mmpL3 in the wild-type, mutant, and complement strains. Changes in gene expression are relative to the appropriate wild-type parent strain control and were calculated using the Pfaffl method [45]. The housekeeping gene sigA was used as the endogenous reference for normalization between samples. Data represent mean ± SEM of three independent biological replicates. Statistical significance was determined using one-way ANOVA (Dunnett’s test) in GraphPad Prism version 10. (*, p = 0.03; ns: not significant). ND: not detected.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nisbett, L.-M.; Previti, M.L.; Seeliger, J.C. A Loss of Function in LprG−Rv1410c Homologues Attenuates Growth during Biofilm Formation in Mycobacterium smegmatis. Pathogens 2023, 12, 1375. https://doi.org/10.3390/pathogens12121375
AMA Style
Nisbett L-M, Previti ML, Seeliger JC. A Loss of Function in LprG−Rv1410c Homologues Attenuates Growth during Biofilm Formation in Mycobacterium smegmatis. Pathogens. 2023; 12(12):1375. https://doi.org/10.3390/pathogens12121375
Chicago/Turabian StyleNisbett, Lisa-Marie, Mary L. Previti, and Jessica C. Seeliger. 2023. "A Loss of Function in LprG−Rv1410c Homologues Attenuates Growth during Biofilm Formation in Mycobacterium smegmatis" Pathogens 12, no. 12: 1375. https://doi.org/10.3390/pathogens12121375
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.