
Inborn Errors of Immunity Predisposing to Herpes Simplex Virus Infections of the Central Nervous System


Abstract
:1. Herpes Simplex Virus Infections of the Central Nervous System
2. Immunity to Herpes Simplex Virus
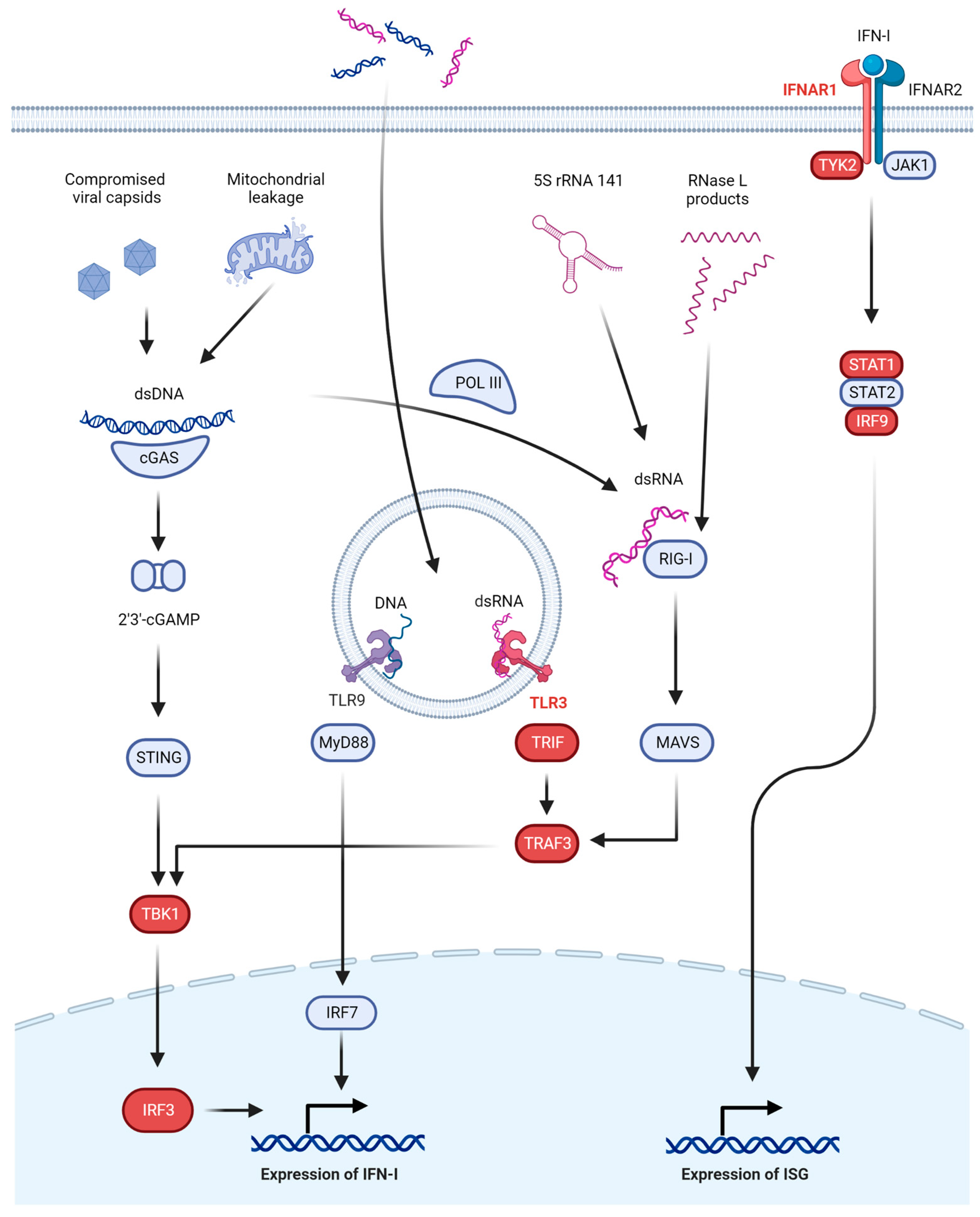
2.1. Interferon-Dependent Anti-HSV Immunity
2.2. Interferon-Independent Anti-HSV Immunity
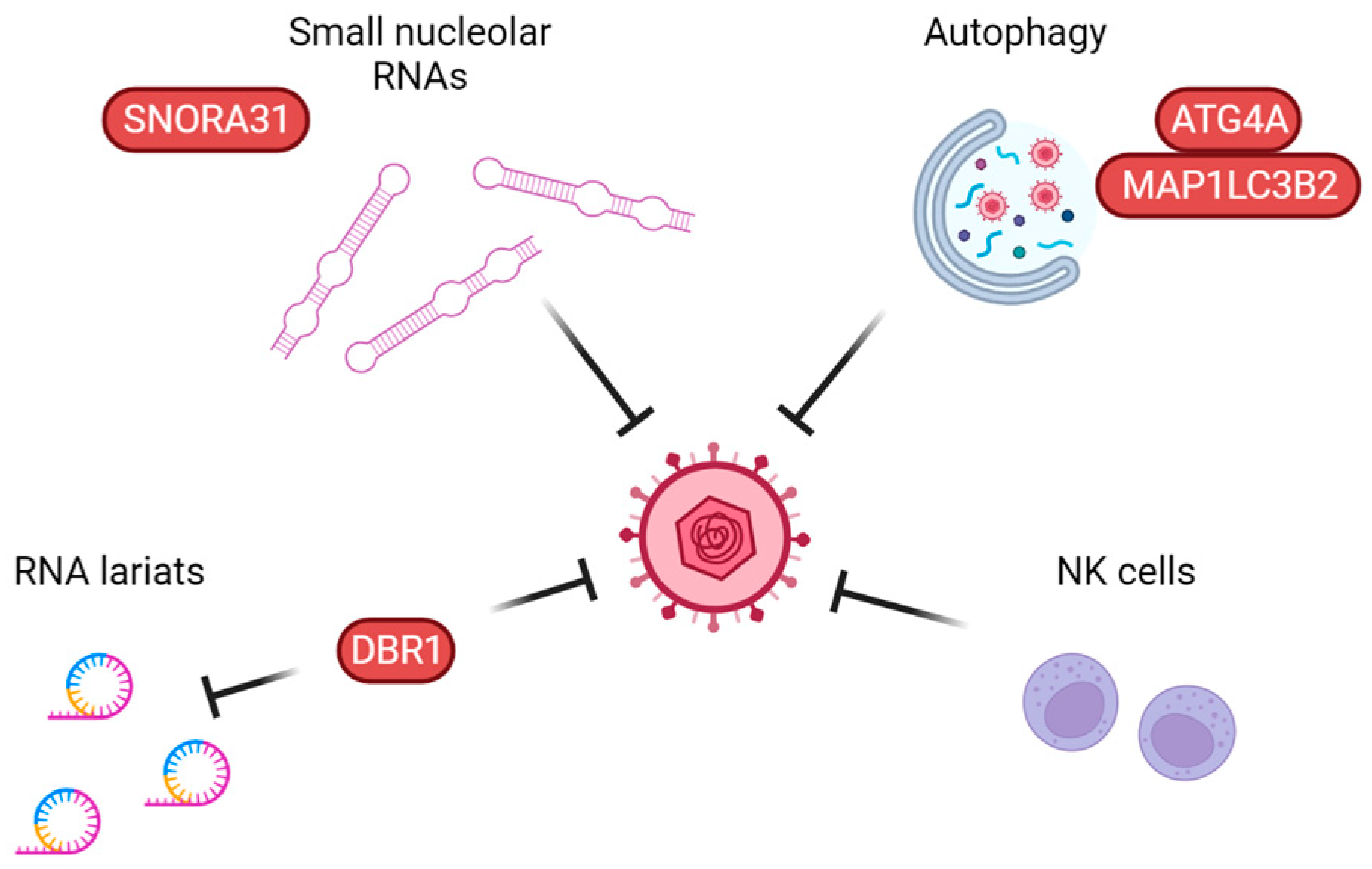
2.2.1. Autophagy
2.2.2. The Lectin Pathway
3. Inborn Errors of Interferon-Dependent Innate Immunity
3.1. TLR3 Signaling
3.2. IFNAR Signaling
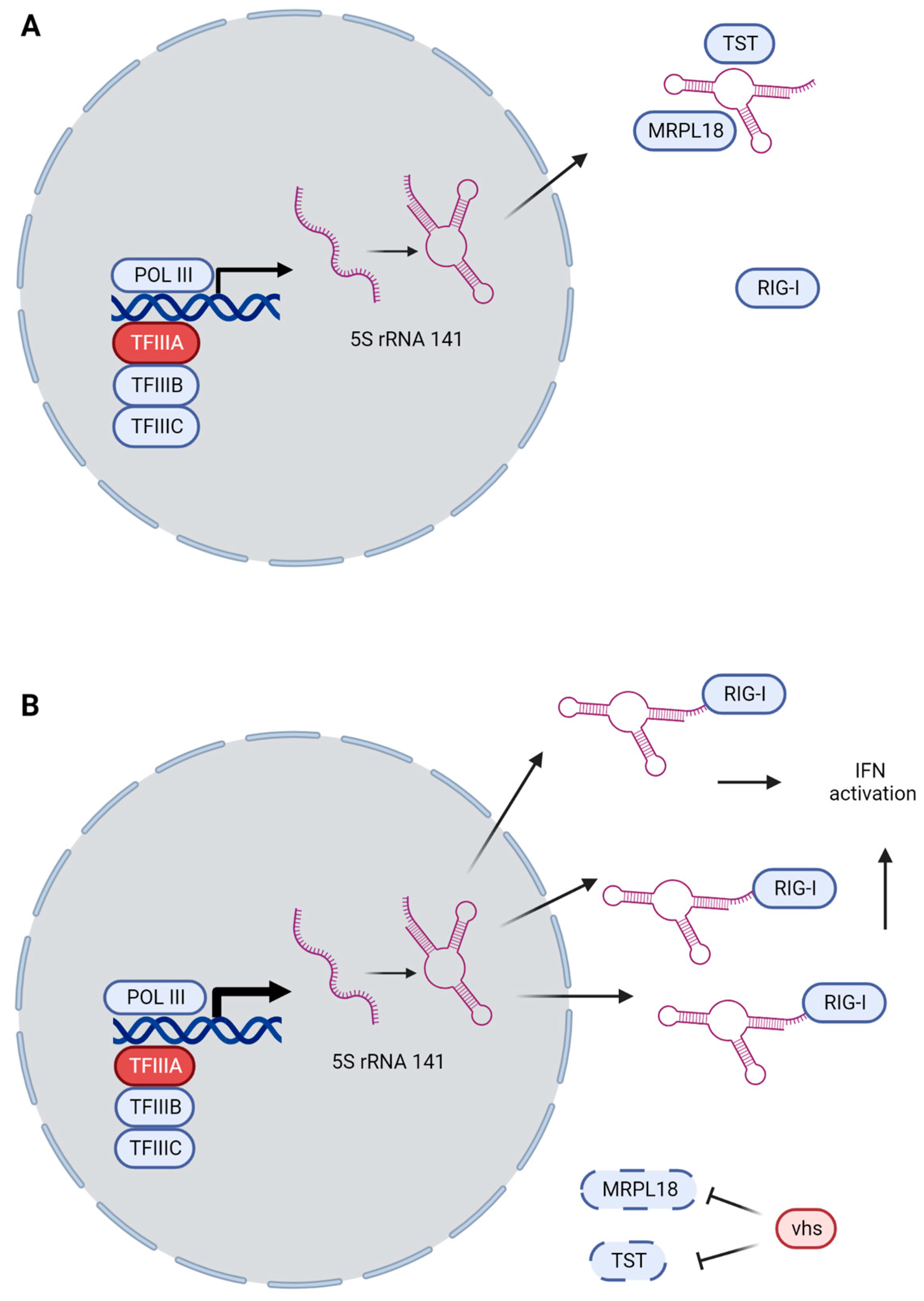
3.3. GTF3A and RNA5SP141 as Guard Mechanisms
4. Inborn Errors of Interferon-Independent Innate and Cell-Intrinsic Immunity
4.1. Autophagy
4.2. Intrinsic Defenses: SNORA31 and DBR1
4.3. Lectin Pathway of Complement Activation
5. Inborn Errors of Lymphocyte Function
6. Other Severe Phenotypes of HSV Infection
7. Perspectives and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wertheim, J.O.; Smith, M.D.; Smith, D.M.; Scheffler, K.; Kosakovsky Pond, S.L. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol. Biol. Evol. 2014, 31, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral Microbiol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Spear, P.G. Infections with herpes simplex viruses (2). N. Engl. J. Med. 1986, 314, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Jmor, F.; Emsley, H.C.; Fischer, M.; Solomon, T.; Lewthwaite, P. The incidence of acute encephalitis syndrome in Western industrialised and tropical countries. Virol. J. 2008, 5, 134. [Google Scholar] [CrossRef]
- Bodilsen, J.; Storgaard, M.; Larsen, L.; Wiese, L.; Helweg-Larsen, J.; Lebech, A.M.; Brandt, C.; Ostergaard, C.; Nielsen, H.; DASGIB Study Group. Infectious meningitis and encephalitis in adults in Denmark: A prospective nationwide observational cohort study (DASGIB). Clin. Microbiol. Infect. 2018, 24, 1102.e1–1102.e5. [Google Scholar] [CrossRef]
- Studahl, M.; Hagberg, L.; Rekabdar, E.; Bergstrom, T. Herpesvirus DNA detection in cerebral spinal fluid: Differences in clinical presentation between alpha-, beta-, and gamma-herpesviruses. Scand. J. Infect. Dis. 2000, 32, 237–248. [Google Scholar] [CrossRef]
- DeBiasi, R.L.; Kleinschmidt-DeMasters, B.K.; Weinberg, A.; Tyler, K.L. Use of PCR for the diagnosis of herpesvirus infections of the central nervous system. J. Clin. Virol. 2002, 25 (Suppl. S1), S5–S11. [Google Scholar] [CrossRef]
- Whitley, R.J.; Alford, C.A.; Hirsch, M.S.; Schooley, R.T.; Luby, J.P.; Aoki, F.Y.; Hanley, D.; Nahmias, A.J.; Soong, S.J. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N. Engl. J. Med. 1986, 314, 144–149. [Google Scholar] [CrossRef]
- Jubelt, B.; Mihai, C.; Li, T.M.; Veerapaneni, P. Rhombencephalitis/brainstem encephalitis. Curr. Neurol. Neurosci. Rep. 2011, 11, 543–552. [Google Scholar] [CrossRef]
- Shukla, B.; Aguilera, E.A.; Salazar, L.; Wootton, S.H.; Kaewpoowat, Q.; Hasbun, R. Aseptic meningitis in adults and children: Diagnostic and management challenges. J. Clin. Virol. 2017, 94, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Jarrin, I.; Sellier, P.; Lopes, A.; Morgand, M.; Makovec, T.; Delcey, V.; Champion, K.; Simoneau, G.; Green, A.; Mouly, S.; et al. Etiologies and Management of Aseptic Meningitis in Patients Admitted to an Internal Medicine Department. Medicine 2016, 95, e2372. [Google Scholar] [CrossRef]
- Mollaret, P. La meningite endothelio-leucocytaire multirecurrente benigne: Syndrome nouveau ou maladie nouvelle? Presentation de deux malades. Bull. Soc. Med. Hop. Paris 1944, 60, 121–122. [Google Scholar]
- Shalabi, M.; Whitley, R.J. Recurrent benign lymphocytic meningitis. Clin. Infect. Dis. 2006, 43, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J.; Matikainen, S.; Paludan, S.R. Activation and evasion of innate antiviral immunity by herpes simplex virus. Viruses 2009, 1, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Verzosa, A.L.; McGeever, L.A.; Bhark, S.J.; Delgado, T.; Salazar, N.; Sanchez, E.L. Herpes Simplex Virus 1 Infection of Neuronal and Non-Neuronal Cells Elicits Specific Innate Immune Responses and Immune Evasion Mechanisms. Front. Immunol. 2021, 12, 644664. [Google Scholar] [CrossRef]
- Whisnant, A.W.; Jürges, C.S.; Hennig, T.; Wyler, E.; Prusty, B.; Rutkowski, A.J.; L’Hernault, A.; Djakovic, L.; Göbel, M.; Döring, K.; et al. Integrative functional genomics decodes herpes simplex virus 1. Nat. Commun. 2020, 11, 2038. [Google Scholar] [CrossRef]
- Dolan, A.; Jamieson, F.E.; Cunningham, C.; Barnett, B.C.; McGeoch, D.J. The genome sequence of herpes simplex virus type 2. J. Virol. 1998, 72, 2010–2021. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Doyle, S.; Vaidya, S.; O’Connell, R.; Dadgostar, H.; Dempsey, P.; Wu, T.; Rao, G.; Sun, R.; Haberland, M.; Modlin, R.; et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 2002, 17, 251–263. [Google Scholar] [CrossRef]
- Cai, M.; Li, M.; Wang, K.; Wang, S.; Lu, Q.; Yan, J.; Mossman, K.L.; Lin, R.; Zheng, C. The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-kappaB through the Toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS ONE 2013, 8, e54586. [Google Scholar] [CrossRef]
- Rasmussen, S.B.; Sorensen, L.N.; Malmgaard, L.; Ank, N.; Baines, J.D.; Chen, Z.J.; Paludan, S.R. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, L.H.; Wilson, S.C.; Morrison, H.M.; Karalis, V.; Chung, J.J.; Chen, K.J.; Bateup, H.S.; Szpara, M.L.; Lee, A.Y.; Cox, J.S.; et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat. Commun. 2020, 11, 3382. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lassig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Bartok, E.; Hartmann, G. Immune Sensing Mechanisms that Discriminate Self from Altered Self and Foreign Nucleic Acids. Immunity 2020, 53, 54–77. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef]
- Kristiansen, H.; Gad, H.H.; Eskildsen-Larsen, S.; Despres, P.; Hartmann, R. The oligoadenylate synthetase family: An ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. 2011, 31, 41–47. [Google Scholar] [CrossRef]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Reinert, L.S.; Lopusna, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vaegter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Bodda, C.; Reinert, L.S.; Fruhwurth, S.; Richardo, T.; Sun, C.; Zhang, B.C.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergstrom, P.; et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217, e20191422. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Esclatine, A. Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”. Viruses 2017, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.T.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef]
- Mauthe, M.; Langereis, M.; Jung, J.; Zhou, X.; Jones, A.; Omta, W.; Tooze, S.A.; Stork, B.; Paludan, S.R.; Ahola, T.; et al. An siRNA screen for ATG protein depletion reveals the extent of the unconventional functions of the autophagy proteome in virus replication. J. Cell Biol. 2016, 214, 619–635. [Google Scholar] [CrossRef]
- Garcia-Laorden, M.I.; Hernandez-Brito, E.; Munoz-Almagro, C.; Pavlovic-Nesic, S.; Rua-Figueroa, I.; Briones, M.L.; Rajas, O.; Borderias, L.; Payeras, A.; Lorente, L.; et al. Should MASP-2 Deficiency Be Considered a Primary Immunodeficiency? Relevance of the Lectin Pathway. J. Clin. Immunol. 2020, 40, 203–210. [Google Scholar] [CrossRef]
- Heja, D.; Harmat, V.; Fodor, K.; Wilmanns, M.; Dobo, J.; Kekesi, K.A.; Zavodszky, P.; Gal, P.; Pal, G. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 Are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J. Biol. Chem. 2012, 287, 20290–20300. [Google Scholar] [CrossRef]
- Gadjeva, M.; Paludan, S.R.; Thiel, S.; Slavov, V.; Ruseva, M.; Eriksson, K.; Lowhagen, G.B.; Shi, L.; Takahashi, K.; Ezekowitz, A.; et al. Mannan-binding lectin modulates the response to HSV-2 infection. Clin. Exp. Immunol. 2004, 138, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Fries, L.F.; Friedman, H.M.; Cohen, G.H.; Eisenberg, R.J.; Hammer, C.H.; Frank, M.M. Glycoprotein C of herpes simplex virus 1 is an inhibitor of the complement cascade. J. Immunol. 1986, 137, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.A.; Randall, R.E.; Hambleton, S. Genetic Lesions of Type I Interferon Signalling in Human Antiviral Immunity. Trends Genet. 2021, 37, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Lim, H.K.; Seppanen, M.; Hautala, T.; Ciancanelli, M.J.; Itan, Y.; Lafaille, F.G.; Dell, W.; Lorenzo, L.; Byun, M.; Pauwels, E.; et al. TLR3 deficiency in herpes simplex encephalitis: High allelic heterogeneity and recurrence risk. Neurology 2014, 83, 1888–1897. [Google Scholar] [CrossRef]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar] [CrossRef]
- Sancho-Shimizu, V.; Perez de Diego, R.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig. 2011, 121, 4889–4902. [Google Scholar] [CrossRef]
- Perez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef]
- Herman, M.; Ciancanelli, M.; Ou, Y.H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; Perez de Diego, R.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef]
- Niehues, T.; Reichenbach, J.; Neubert, J.; Gudowius, S.; Puel, A.; Horneff, G.; Lainka, E.; Dirksen, U.; Schroten, H.; Doffinger, R.; et al. Nuclear factor kappaB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J. Allergy Clin. Immunol. 2004, 114, 1456–1462. [Google Scholar] [CrossRef]
- Puel, A.; Reichenbach, J.; Bustamante, J.; Ku, C.L.; Feinberg, J.; Doffinger, R.; Bonnet, M.; Filipe-Santos, O.; de Beaucoudrey, L.; Durandy, A.; et al. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am. J. Hum. Genet. 2006, 78, 691–701. [Google Scholar] [CrossRef]
- Audry, M.; Ciancanelli, M.; Yang, K.; Cobat, A.; Chang, H.H.; Sancho-Shimizu, V.; Lorenzo, L.; Niehues, T.; Reichenbach, J.; Li, X.X.; et al. NEMO is a key component of NF-kappaB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J. Allergy Clin. Immunol. 2011, 128, 610–617. [Google Scholar] [CrossRef]
- Andersen, L.L.; Mork, N.; Reinert, L.S.; Kofod-Olsen, E.; Narita, R.; Jorgensen, S.E.; Skipper, K.A.; Honing, K.; Gad, H.H.; Ostergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef]
- Naesens, L.; Muppala, S.; Acharya, D.; Nemegeer, J.; Bogaert, D.; Lee, J.H.; Staes, K.; Debacker, V.; De Bleser, P.; De Bruyne, M.; et al. GTF3A mutations predispose to herpes simplex encephalitis by disrupting biogenesis of the host-derived RIG-I ligand RNA5SP141. Sci. Immunol. 2022, 7, eabq4531. [Google Scholar] [CrossRef]
- Bastard, P.; Manry, J.; Chen, J.; Rosain, J.; Seeleuthner, Y.; AbuZaitun, O.; Lorenzo, L.; Khan, T.; Hasek, M.; Hernandez, N.; et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J. Clin. Investig. 2021, 131, e139980. [Google Scholar] [CrossRef]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Bravo Garcia-Morato, M.; Calvo Apalategi, A.; Bravo-Gallego, L.Y.; Blazquez Moreno, A.; Simon-Fuentes, M.; Garmendia, J.V.; Mendez Echevarria, A.; Del Rosal Rabes, T.; Dominguez-Soto, A.; Lopez-Granados, E.; et al. Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J. Allergy Clin. Immunol. 2019, 144, 309–312.e310. [Google Scholar] [CrossRef]
- Lafaille, F.G.; Harschnitz, O.; Lee, Y.S.; Zhang, P.; Hasek, M.L.; Kerner, G.; Itan, Y.; Ewaleifoh, O.; Rapaport, F.; Carlile, T.M.; et al. Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat. Med. 2019, 25, 1873–1884. [Google Scholar] [CrossRef]
- Bibert, S.; Piret, J.; Quinodoz, M.; Collinet, E.; Zoete, V.; Michielin, O.; Menasria, R.; Meylan, P.; Bihl, T.; Erard, V.; et al. Herpes simplex encephalitis in adult patients with MASP-2 deficiency. PLoS Pathog. 2019, 15, e1008168. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Clark, N.E.; Freije, C.A.; Pauwels, E.; Taggart, A.J.; Okada, S.; Mandel, H.; Garcia, P.; Ciancanelli, M.J.; Biran, A.; et al. Inborn Errors of RNA Lariat Metabolism in Humans with Brainstem Viral Infection. Cell 2018, 172, 952–965.e918. [Google Scholar] [CrossRef]
- Kreins, A.Y.; Ciancanelli, M.J.; Okada, S.; Kong, X.F.; Ramirez-Alejo, N.; Kilic, S.S.; El Baghdadi, J.; Nonoyama, S.; Mahdaviani, S.A.; Ailal, F.; et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J. Exp. Med. 2015, 212, 1641–1662. [Google Scholar] [CrossRef]
- Hait, A.S.; Olagnier, D.; Sancho-Shimizu, V.; Skipper, K.A.; Helleberg, M.; Larsen, S.M.; Bodda, C.; Moldovan, L.I.; Ren, F.; Brinck Andersen, N.S.; et al. Defects in LC3B2 and ATG4A underlie HSV2 meningitis and reveal a critical role for autophagy in antiviral defense in humans. Sci. Immunol. 2020, 5, 54. [Google Scholar] [CrossRef]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef]
- Gao, D.; Ciancanelli, M.J.; Zhang, P.; Harschnitz, O.; Bondet, V.; Hasek, M.; Chen, J.; Mu, X.; Itan, Y.; Cobat, A.; et al. TLR3 controls constitutive IFN-beta antiviral immunity in human fibroblasts and cortical neurons. J. Clin. Investig. 2021, 131, eabc2691. [Google Scholar] [CrossRef]
- Hernandez, N.; Bucciol, G.; Moens, L.; Le Pen, J.; Shahrooei, M.; Goudouris, E.; Shirkani, A.; Changi-Ashtiani, M.; Rokni-Zadeh, H.; Sayar, E.H.; et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J. Exp. Med. 2019, 216, 2057–2070. [Google Scholar] [CrossRef]
- Meyts, I. Null IFNAR1 and IFNAR2 alleles are surprisingly common in the Pacific and Arctic. J. Exp. Med. 2022, 219, e20220491. [Google Scholar] [CrossRef]
- Duncan, C.J.; Mohamad, S.M.; Young, D.F.; Skelton, A.J.; Leahy, T.R.; Munday, D.C.; Butler, K.M.; Morfopoulou, S.; Brown, J.R.; Hubank, M.; et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci. Transl. Med. 2015, 7, 307ra154. [Google Scholar] [CrossRef]
- Duncan, C.J.A.; Skouboe, M.K.; Howarth, S.; Hollensen, A.K.; Chen, R.; Borresen, M.L.; Thompson, B.J.; Stremenova Spegarova, J.; Hatton, C.F.; Staeger, F.F.; et al. Life-threatening viral disease in a novel form of autosomal recessive IFNAR2 deficiency in the Arctic. J. Exp. Med. 2022, 219, e20212427. [Google Scholar] [CrossRef]
- Dupuis, S.; Dargemont, C.; Fieschi, C.; Thomassin, N.; Rosenzweig, S.; Harris, J.; Holland, S.M.; Schreiber, R.D.; Casanova, J.L. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 2001, 293, 300–303. [Google Scholar] [CrossRef]
- Le Voyer, T.; Sakata, S.; Tsumura, M.; Khan, T.; Esteve-Sole, A.; Al-Saud, B.K.; Gungor, H.E.; Taur, P.; Jeanne-Julien, V.; Christiansen, M.; et al. Genetic, Immunological, and Clinical Features of 32 Patients with Autosomal Recessive STAT1 Deficiency. J. Immunol. 2021, 207, 133–152. [Google Scholar] [CrossRef]
- Sarrafzadeh, S.A.; Mahloojirad, M.; Casanova, J.L.; Badalzadeh, M.; Bustamante, J.; Boisson-Dupuis, S.; Pourpak, Z.; Nourizadeh, M.; Moin, M. A New Patient with Inherited TYK2 Deficiency. J. Clin. Immunol. 2020, 40, 232–235. [Google Scholar] [CrossRef]
- Mork, N.; Kofod-Olsen, E.; Sorensen, K.B.; Bach, E.; Orntoft, T.F.; Ostergaard, L.; Paludan, S.R.; Christiansen, M.; Mogensen, T.H. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. 2015, 16, 552–566. [Google Scholar] [CrossRef]
- Van der Biezen, E.A.; Jones, J.D. Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem. Sci. 1998, 23, 454–456. [Google Scholar] [CrossRef]
- Borish, L.; Ayars, A.G.; Kirkpatrick, C.H. Common variable immunodeficiency presenting as herpes simplex virus encephalitis. J. Allergy Clin. Immunol. 2011, 127, 541–543. [Google Scholar] [CrossRef]
- Paludan, S.R.; Mogensen, T.H. Constitutive and latent immune mechanisms exert ‘silent’ control of virus infections in the central nervous system. Curr. Opin. Immunol. 2021, 72, 158–166. [Google Scholar] [CrossRef]
- Tang, S.; Patel, A.; Krause, P.R. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc. Natl. Acad. Sci. USA 2016, 113, 12256–12261. [Google Scholar] [CrossRef]
- Seppanen, M.; Lokki, M.L.; Lappalainen, M.; Hiltunen-Back, E.; Rovio, A.T.; Kares, S.; Hurme, M.; Aittoniemi, J. Mannose-binding lectin 2 gene polymorphism in recurrent herpes simplex virus 2 infection. Hum. Immunol. 2009, 70, 218–221. [Google Scholar] [CrossRef]
- Stengaard-Pedersen, K.; Thiel, S.; Gadjeva, M.; Moller-Kristensen, M.; Sorensen, R.; Jensen, L.T.; Sjoholm, A.G.; Fugger, L.; Jensenius, J.C. Inherited deficiency of mannan-binding lectin-associated serine protease 2. N. Engl. J. Med. 2003, 349, 554–560. [Google Scholar] [CrossRef]
- Notarangelo, L.; Casanova, J.L.; Fischer, A.; Puck, J.; Rosen, F.; Seger, R.; Geha, R. International Union of Immunological Societies Primary Immunodeficiency diseases classification, c. Primary immunodeficiency diseases: An update. J. Allergy Clin. Immunol. 2004, 114, 677–687. [Google Scholar] [CrossRef]
- Verdu, P.; Barreiro, L.B.; Patin, E.; Gessain, A.; Cassar, O.; Kidd, J.R.; Kidd, K.K.; Behar, D.M.; Froment, A.; Heyer, E.; et al. Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum. Mol. Genet. 2006, 15, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.L.; Abel, L. Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin. Immunol. 2018, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef] [PubMed]
- Jouanguy, E.; Beziat, V.; Mogensen, T.H.; Casanova, J.L.; Tangye, S.G.; Zhang, S.Y. Human inborn errors of immunity to herpes viruses. Curr. Opin. Immunol. 2020, 62, 106–122. [Google Scholar] [CrossRef]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef]
- Orange, J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef]
- Mace, E.M.; Hsu, A.P.; Monaco-Shawver, L.; Makedonas, G.; Rosen, J.B.; Dropulic, L.; Cohen, J.I.; Frenkel, E.P.; Bagwell, J.C.; Sullivan, J.L.; et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood 2013, 121, 2669–2677. [Google Scholar] [CrossRef]
- Almerigogna, F.; Fassio, F.; Giudizi, M.G.; Biagiotti, R.; Manuelli, C.; Chiappini, E.; Galli, L.; Romagnani, S.; De Martino, M. Natural killer cell deficiencies in a consecutive series of children with herpetic encephalitis. Int. J. Immunopathol. Pharmacol. 2011, 24, 231–238. [Google Scholar] [CrossRef]
- Lisco, A.; Hsu, A.P.; Dimitrova, D.; Proctor, D.M.; Mace, E.M.; Ye, P.; Anderson, M.V.; Hicks, S.N.; Grivas, C.; Hammoud, D.A.; et al. Treatment of Relapsing HPV Diseases by Restored Function of Natural Killer Cells. N. Engl. J. Med. 2021, 385, 921–929. [Google Scholar] [CrossRef]
- Salzer, U.; Warnatz, K.; Peter, H.H. Common variable immunodeficiency: An update. Arthritis Res. Ther. 2012, 14, 223. [Google Scholar] [CrossRef]
- Lourdes, L.S.; Daily, K.C. Common variable immunodeficiency syndrome in an adult. Lancet 2014, 383, 926. [Google Scholar] [CrossRef]
- Cummings, L.; Tucker, M.; Gibson, M.; Myers, A.; Pastinen, T.; Johnston, J.; Farrow, E.; Sampath, V. Rare Genetic Variants in Immune Genes and Neonatal Herpes Simplex Viral Infections. Pediatrics 2021, 147, e20200687. [Google Scholar] [CrossRef]
- Hodara, E.; Ong, P.Y. The Genetics of Eczema Herpeticum. Clin. Rev. Allergy Immunol. 2022, 63, 390–397. [Google Scholar] [CrossRef]
- Bin, L.; Malley, C.; Taylor, P.; Preethi Boorgula, M.; Chavan, S.; Daya, M.; Mathias, M.; Shankar, G.; Rafaels, N.; Vergara, C.; et al. Whole genome sequencing identifies novel genetic mutations in patients with eczema herpeticum. Allergy 2021, 76, 2510–2523. [Google Scholar] [CrossRef]
- Fox, L.E.; Locke, M.C.; Lenschow, D.J. Context Is Key: Delineating the Unique Functions of IFNα and IFNβ in Disease. Front. Immunol. 2020, 11, 606874. [Google Scholar] [CrossRef]
- Gao, L.; Bin, L.; Rafaels, N.M.; Huang, L.; Potee, J.; Ruczinski, I.; Beaty, T.H.; Paller, A.S.; Schneider, L.C.; Gallo, R.; et al. Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J. Allergy Clin. Immunol. 2015, 136, 1591–1600. [Google Scholar] [CrossRef]
- Gao, P.S.; Rafaels, N.M.; Hand, T.; Murray, T.; Boguniewicz, M.; Hata, T.; Schneider, L.; Hanifin, J.M.; Gallo, R.L.; Gao, L.; et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J. Allergy Clin. Immunol. 2009, 124, 507–513.e7. [Google Scholar] [CrossRef]
- Eaaswarkhanth, M.; Xu, D.; Flanagan, C.; Rzhetskaya, M.; Hayes, M.G.; Blekhman, R.; Jablonski, N.G.; Gokcumen, O. Atopic Dermatitis Susceptibility Variants in Filaggrin Hitchhike Hornerin Selective Sweep. Genome Biol. Evol. 2016, 8, 3240–3255. [Google Scholar] [CrossRef]
- Kim, B.E.; Bin, L.; Ye, Y.M.; Ramamoorthy, P.; Leung, D.Y.M. IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J. Investig. Dermatol. 2013, 133, 2678–2685. [Google Scholar] [CrossRef]
- Ishii, E. Hemophagocytic Lymphohistiocytosis in Children: Pathogenesis and Treatment. Front. Pediatr. 2016, 4, 47. [Google Scholar] [CrossRef]
- Canna, S.W.; Marsh, R.A. Pediatric hemophagocytic lymphohistiocytosis. Blood 2020, 135, 1332–1343. [Google Scholar] [CrossRef]
- McKeone, D.J.; DeMartini, T.K.M.; Kavanagh, R.P.; Halstead, E.S. Case Report: Rapid Recognition and Immune Modulation of Secondary HLH Due to Disseminated HSV Infection. Front. Pediatr. 2021, 9, 681055. [Google Scholar] [CrossRef]
- Takehara, H.; Hirohata, K.; Mutoh, H.; Irisa, C.; Kakiuchi, S.; Nishimura, R.; Oka, A.; Takahashi, N. Critically Severe Case of Neonatal Herpes with High Viral Load and Hemophagocytic Syndrome. Tohoku J. Exp. Med. 2019, 247, 149–152. [Google Scholar] [CrossRef]
- States, V.A.; Kapp, M.E. Herpes simplex virus-1 triggered hemophagocytic lymphohistiocytosis in a patient with granulomatosis with polyangiitis. Autops. Case Rep. 2022, 12, e2021395. [Google Scholar] [CrossRef]
- Yabushita, T.; Yoshioka, S.; Koba, Y.; Ono, Y.; Hiramoto, N.; Tabata, S.; Itou, M.; Shimizu, N.; Tomii, K.; Ishikawa, T. Successful Treatment of Herpes Simplex Virus (HSV)-1-associated Hemophagocytic Lymphohistiocytosis (HLH) with Acyclovir: A Case Report and Literature Review. Intern. Med. 2017, 56, 2919–2923. [Google Scholar] [CrossRef]
- Yamada, K.; Yamamoto, Y.; Uchiyama, A.; Ito, R.; Aoki, Y.; Uchida, Y.; Nagasawa, H.; Kimura, H.; Ichiyama, T.; Fukao, T.; et al. Successful treatment of neonatal herpes simplex-type 1 infection complicated by hemophagocytic lymphohistiocytosis and acute liver failure. Tohoku J. Exp. Med. 2008, 214, 1–5. [Google Scholar] [CrossRef]
- Kurosawa, S.; Sekiya, N.; Fukushima, K.; Ikeuchi, K.; Fukuda, A.; Takahashi, H.; Chen, F.; Hasegawa, H.; Katano, H.; Hishima, T.; et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect. Dis. 2019, 19, 65. [Google Scholar] [CrossRef]
- Freytag, M.R.; Jorgensen, S.E.; Thomsen, M.M.; Al-Mousawi, A.; Hait, A.S.; Olagnier, D.; Bay, J.T.; Helleberg, M.; Mogensen, T.H. Postpartum Disseminated Herpes Simplex Virus Type 1 Infection With Hemophagocytic Lymphohistiocytosis and Fulminant Neonatal Herpes Infection. J. Infect. Dis. 2022, 225, 157–162. [Google Scholar] [CrossRef]
- Saettini, F.; Radaelli, S.; Ocello, L.; Ferrari, G.M.; Corti, P.; Dell’Acqua, F.; Ippolito, D.; Foresti, S.; Gervasini, C.; Badolato, R.; et al. Secondary hemophagocytic lymphohystiocytosis in a Rubinstein Taybi syndrome patient. Pediatr. Hematol. Oncol. 2022, 39, 74–79. [Google Scholar] [CrossRef]
- Spinner, M.A.; Ker, J.P.; Stoudenmire, C.J.; Fadare, O.; Mace, E.M.; Orange, J.S.; Hsu, A.P.; Holland, S.M. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J. Allergy Clin. Immunol. 2016, 137, 638–640. [Google Scholar] [CrossRef]
- Casanova, J.L. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc. Natl. Acad. Sci. USA 2015, 112, E7128–E7137. [Google Scholar] [CrossRef]
- Casanova, J.L.; Conley, M.E.; Seligman, S.J.; Abel, L.; Notarangelo, L.D. Guidelines for genetic studies in single patients: Lessons from primary immunodeficiencies. J. Exp. Med. 2014, 211, 2137–2149. [Google Scholar] [CrossRef]
- Casanova, J.L.; Abel, L. From rare disorders of immunity to common determinants of infection: Following the mechanistic thread. Cell 2022, 185, 3086–3103. [Google Scholar] [CrossRef]
- Gros, P.; Casanova, J.L. Reconciling Mouse and Human Immunology at the Altar of Genetics. Annu. Rev. Immunol. 2022, 219, e20220028. [Google Scholar] [CrossRef]
- Bastard, P.; Hsiao, K.C.; Zhang, Q.; Choin, J.; Best, E.; Chen, J.; Gervais, A.; Bizien, L.; Materna, M.; Harmant, C.; et al. A loss-of-function IFNAR1 allele in Polynesia underlies severe viral diseases in homozygotes. J. Exp. Med. 2022, 219, e20220028. [Google Scholar] [CrossRef]
- Gough, D.J.; Messina, N.L.; Clarke, C.J.; Johnstone, R.W.; Levy, D.E. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 2012, 36, 166–174. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef]
- Brinck Andersen, N.S.; Jorgensen, S.E.; Skipper, K.A.; Larsen, S.M.; Heinz, J.; Thomsen, M.M.; Farahani, E.; Cai, Y.; Hait, A.S.; Kay, L.; et al. Essential role of autophagy in restricting poliovirus infection revealed by identification of an ATG7 defect in a poliomyelitis patient. Autophagy 2021, 17, 2449–2464. [Google Scholar] [CrossRef]
- Paludan, S.R.; Pradeu, T.; Masters, S.L.; Mogensen, T.H. Constitutive immune mechanisms: Mediators of host defence and immune regulation. Nat. Rev. Immunol. 2021, 21, 137–150. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Signaling Pathway Affected | Gene | Variants | References |
|---|---|---|---|---|
| HSE | TLR3 | UNC93B1 | c.1034del4 | [64] |
| c.781G>A | ||||
| TLR3 | p.P554S | [65,66,67] | ||
| p.E746X | ||||
| p.G743D | ||||
| p.R811I | ||||
| p.D592N | ||||
| p.M374T | ||||
| p.L360P | ||||
| p.R867Q | ||||
| TRIF | p.R141X | [68] | ||
| p.S186L | ||||
| TLR3/RIG-I | TRAF3 | p. R118W | [69] | |
| TLR3/RIG-I/STING | TBK1 | p.G159A | [70] | |
| p.D50A | ||||
| NEMO | p.M38fsX48 | [71,72,73] | ||
| IRF3 | p.R285Q | [74] | ||
| 5S rRNA 141/RIG-I | GTF3A | p.C195W | [75] | |
| p.C219R | ||||
| IFNAR signaling | IFNAR1 | g.34,726,420_34,728,094del | [76] | |
| STAT1 | c.1757–1758delAG | [77] | ||
| IRF9 | c.577+1G>T | [78] | ||
| Small nucleolar RNA | SNORA31 | n.36T>C | [79] | |
| n.75C>G | ||||
| n.96T>G | ||||
| n.111T>C | ||||
| Lectin pathway * | MASP2 | p.R203W | [80] | |
| p.G634R | ||||
| Brainstem encephalitis | RNA lariat metabolism | DBR1 | p.I120T | [81] |
| Meningitis | IFNAR signaling | TYK2 | p.R638X | [82] |
| Mollaret’s meningitis | Autophagy * | ATG4A | p.L90I | [83] |
| MAP1LC3B2 | p.L109M | [83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skouboe, M.K.; Werner, M.; Mogensen, T.H. Inborn Errors of Immunity Predisposing to Herpes Simplex Virus Infections of the Central Nervous System. Pathogens 2023, 12, 310. https://doi.org/10.3390/pathogens12020310
Skouboe MK, Werner M, Mogensen TH. Inborn Errors of Immunity Predisposing to Herpes Simplex Virus Infections of the Central Nervous System. Pathogens. 2023; 12(2):310. https://doi.org/10.3390/pathogens12020310
Chicago/Turabian StyleSkouboe, Morten Kelder, Marvin Werner, and Trine H. Mogensen. 2023. "Inborn Errors of Immunity Predisposing to Herpes Simplex Virus Infections of the Central Nervous System" Pathogens 12, no. 2: 310. https://doi.org/10.3390/pathogens12020310