

Early Transcriptional Responses of Human Nasal Epithelial Cells to Infection with Influenza A and SARS-CoV-2 Virus Differ and Are Influenced by Physiological Temperature



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. Virus Seed Stock and Working Stock Generation
2.3. TCID50 Assay
2.4. Multiplicity of Infection (MOI) Infections
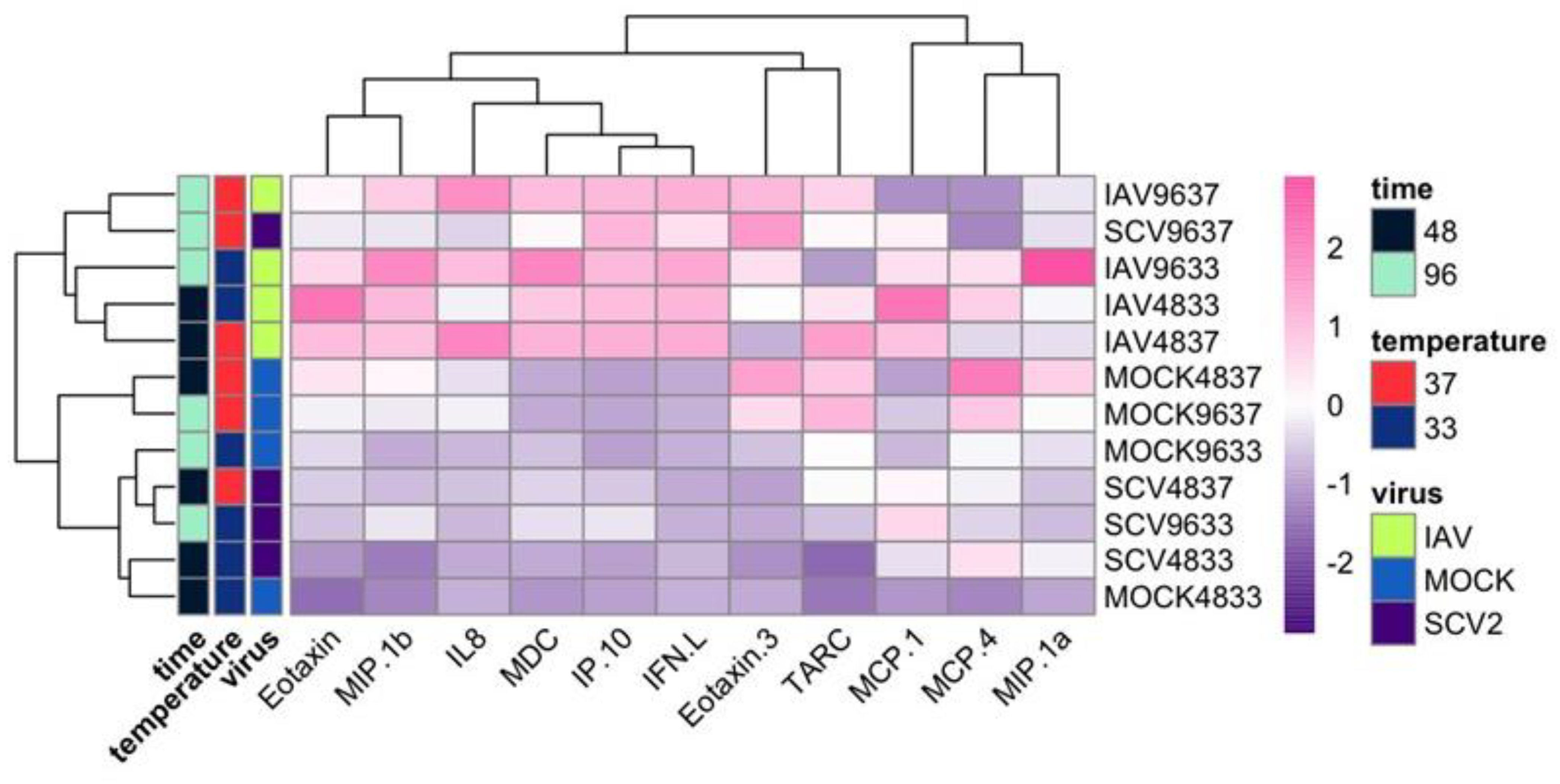
2.5. Cytokine Secretion
2.6. RNA-Sequencing and Analysis
3. Results
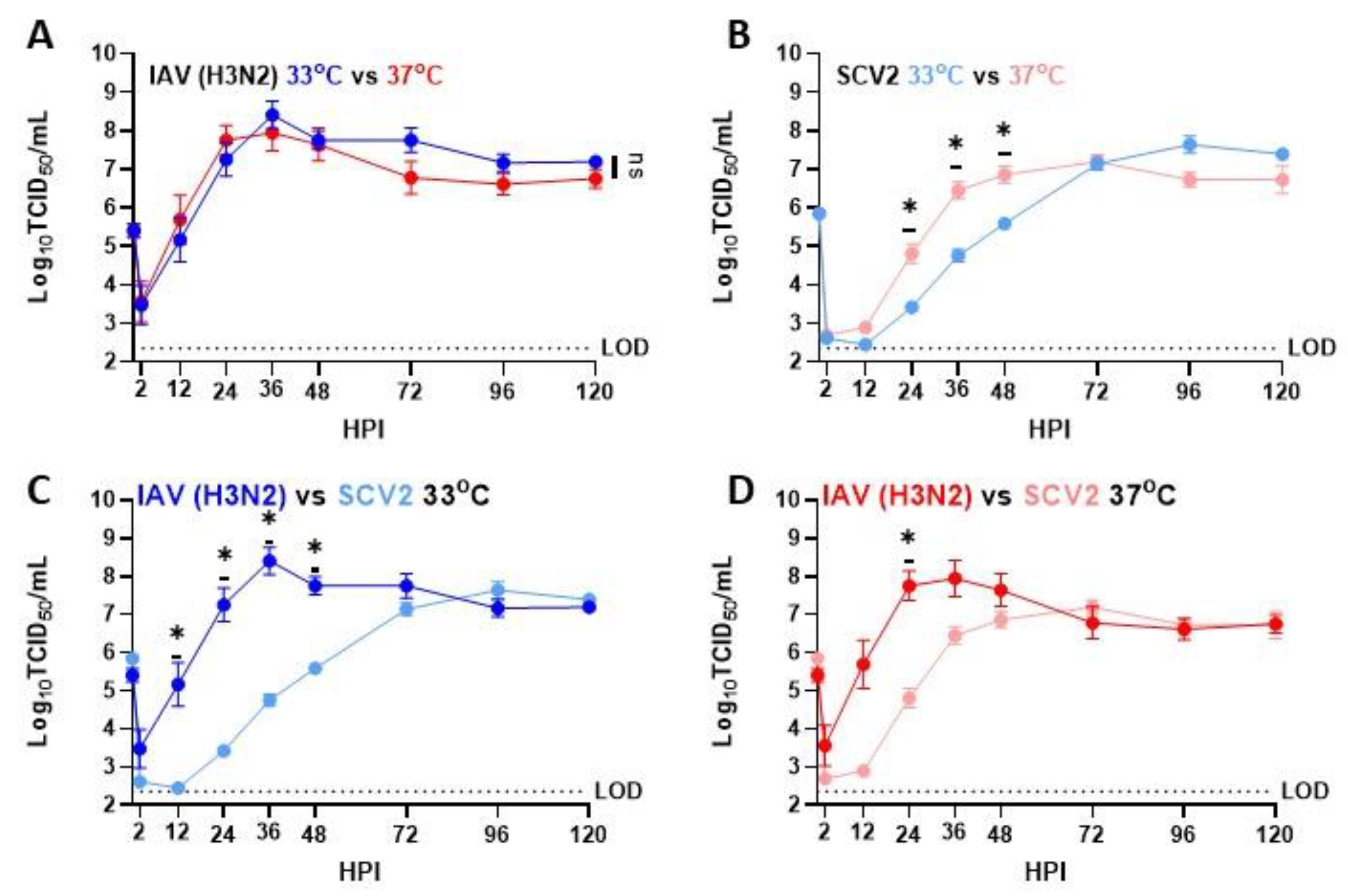
3.1. Temperature-Dependent Replication in IAV and SCV2 on Human Nasal Epithelial Cells
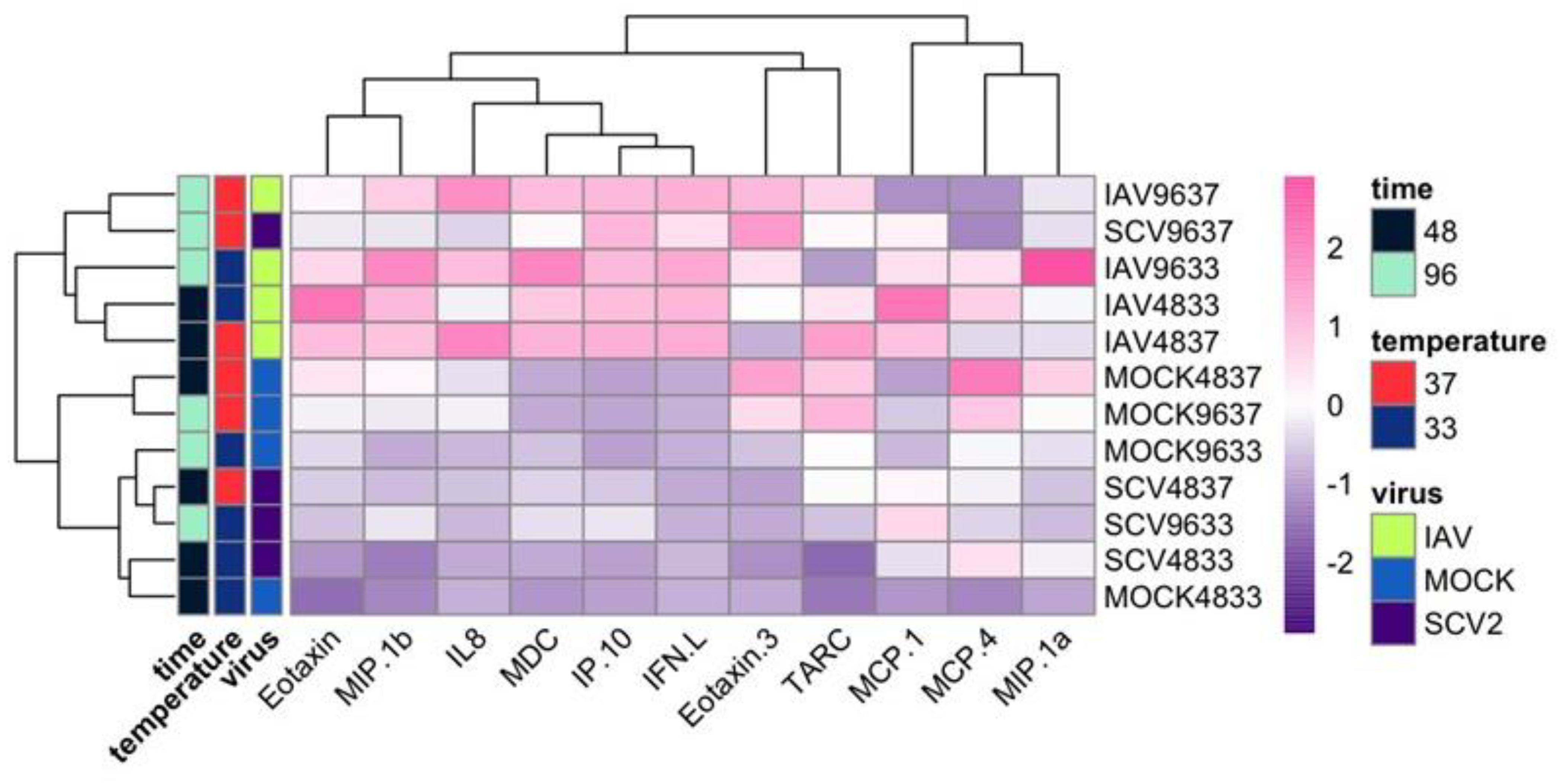
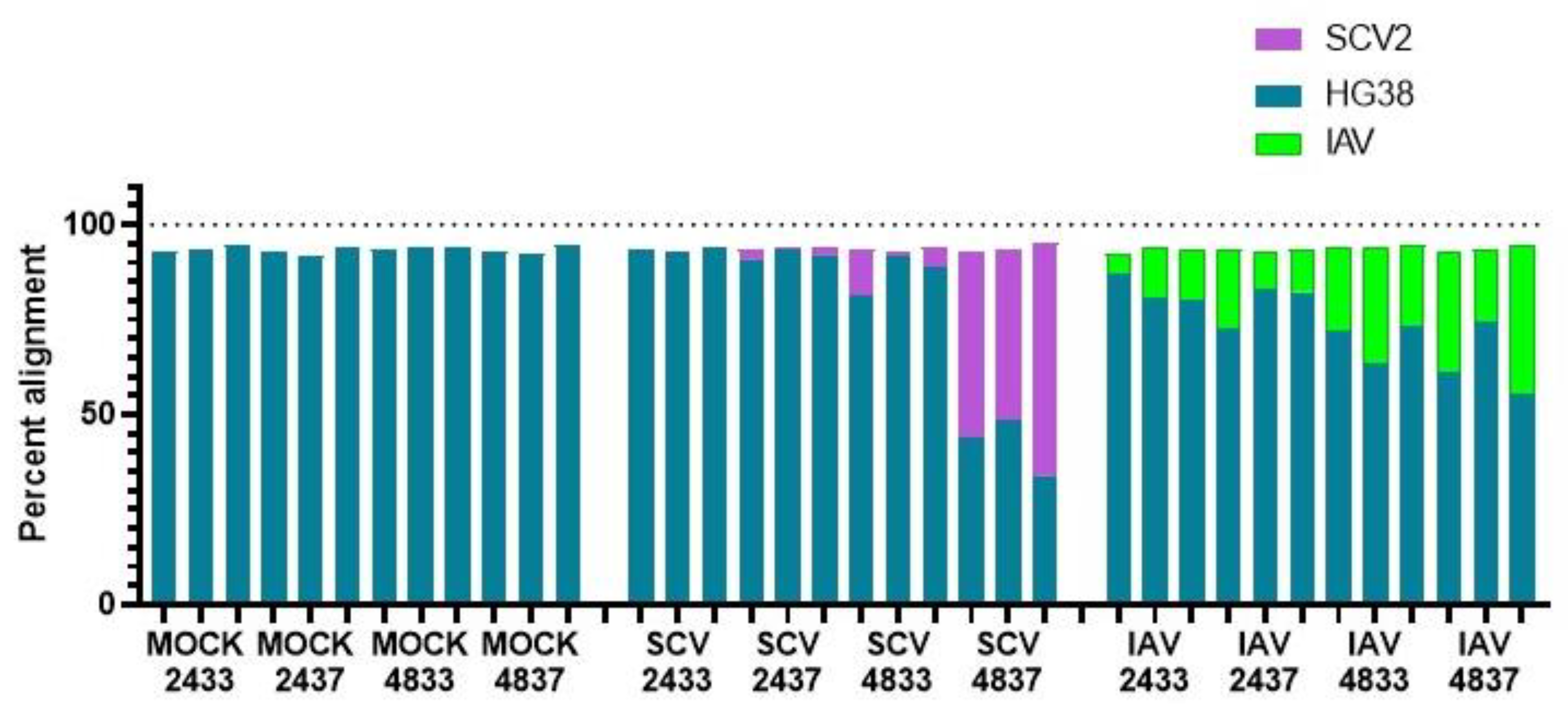
3.2. RNA-Seq of Infected hNECs at Different Physiological Temperatures
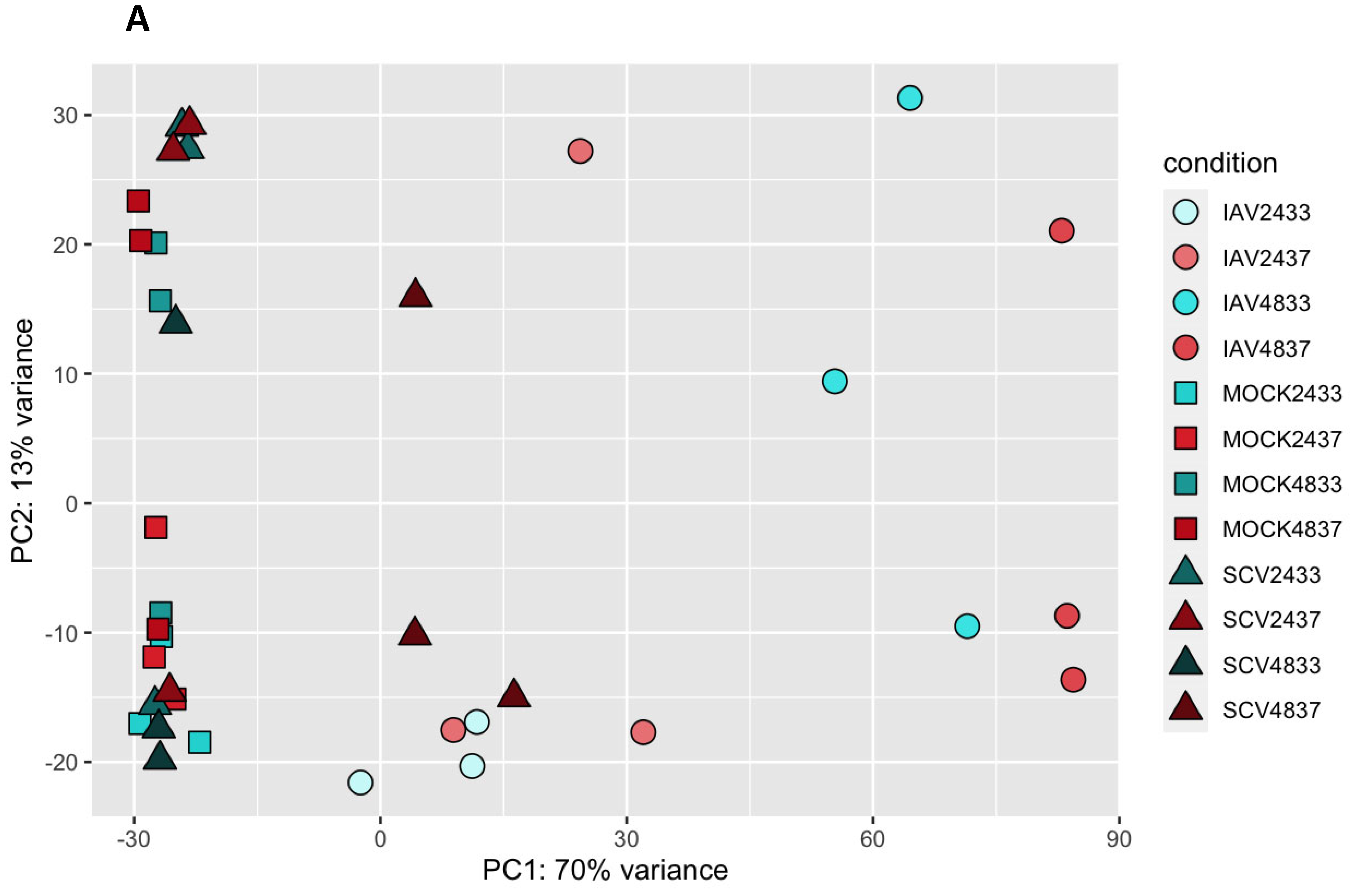
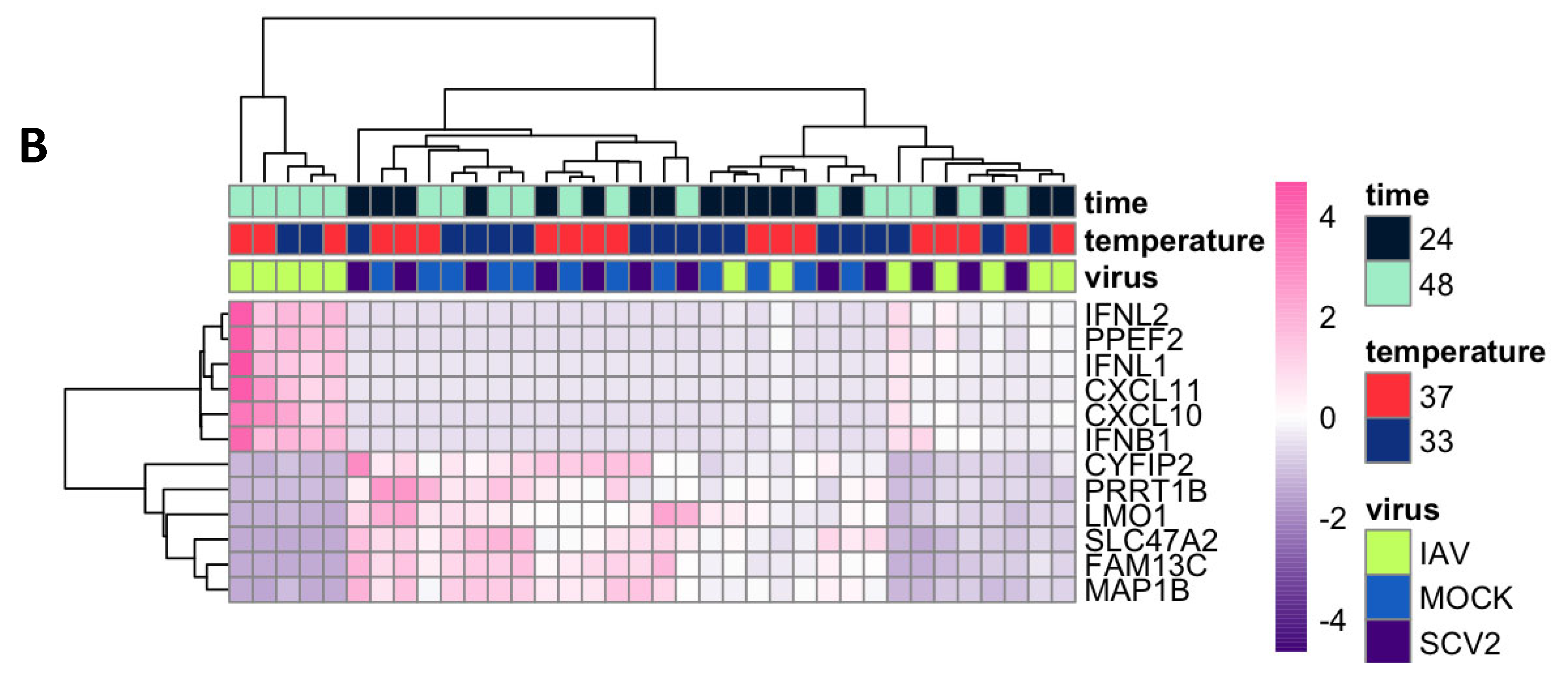
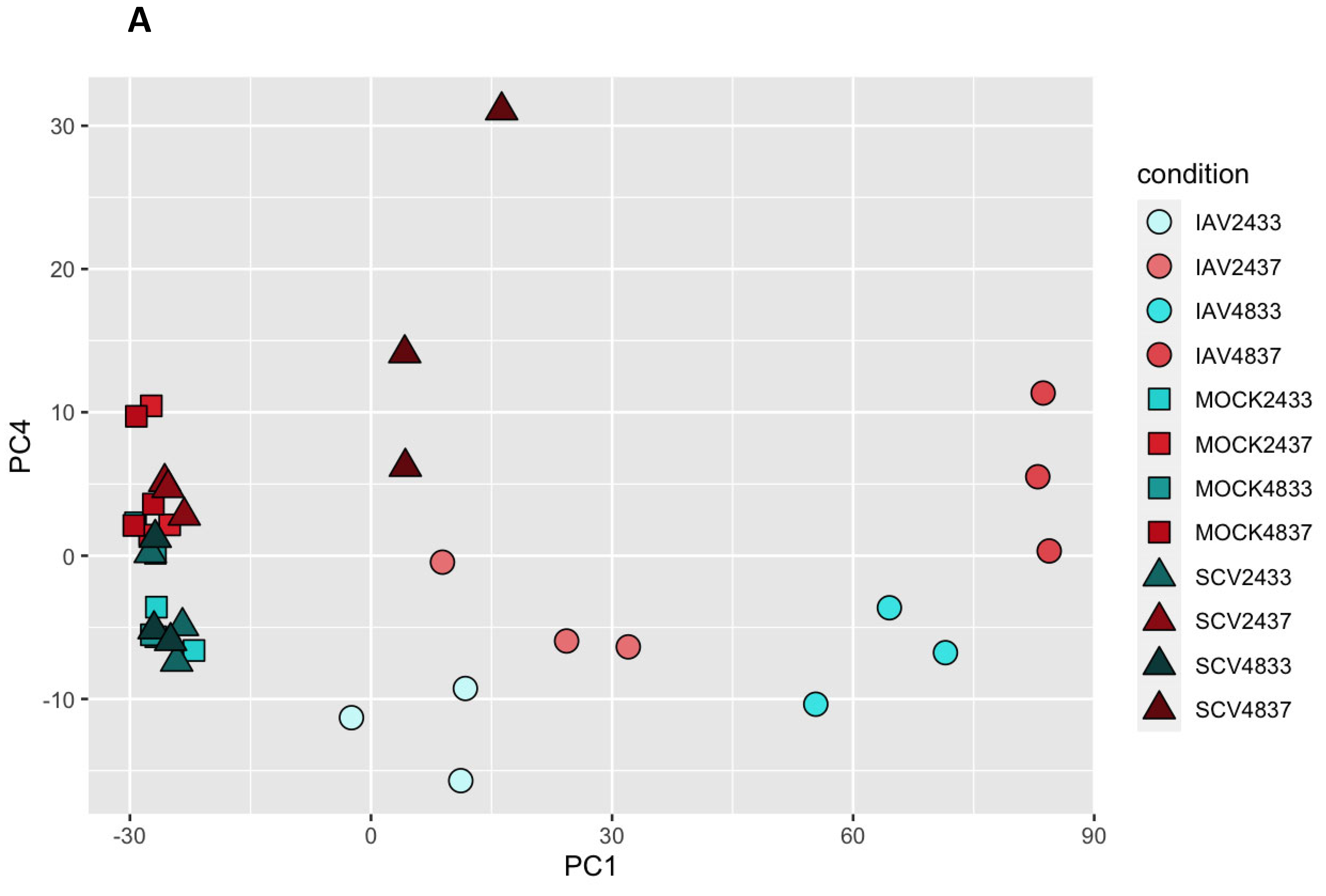
3.3. Variance in Dataset Due to Infection and Temperature
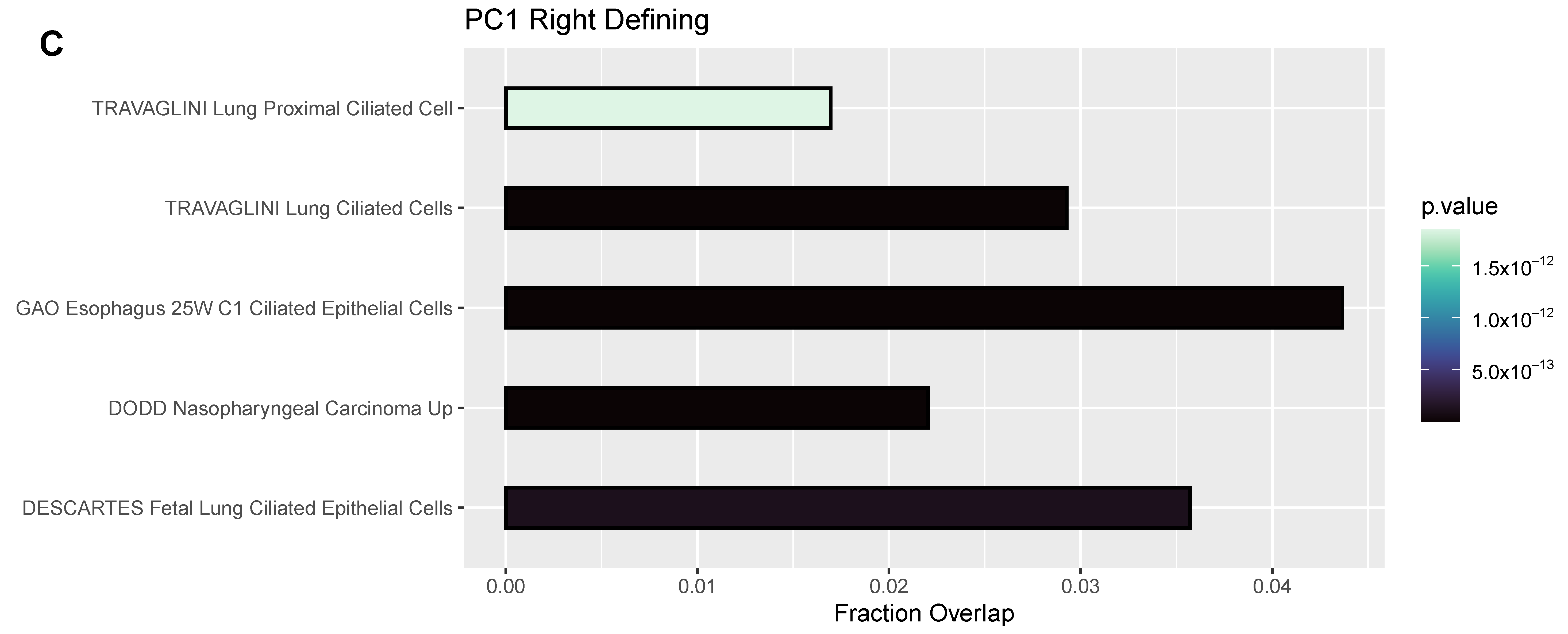
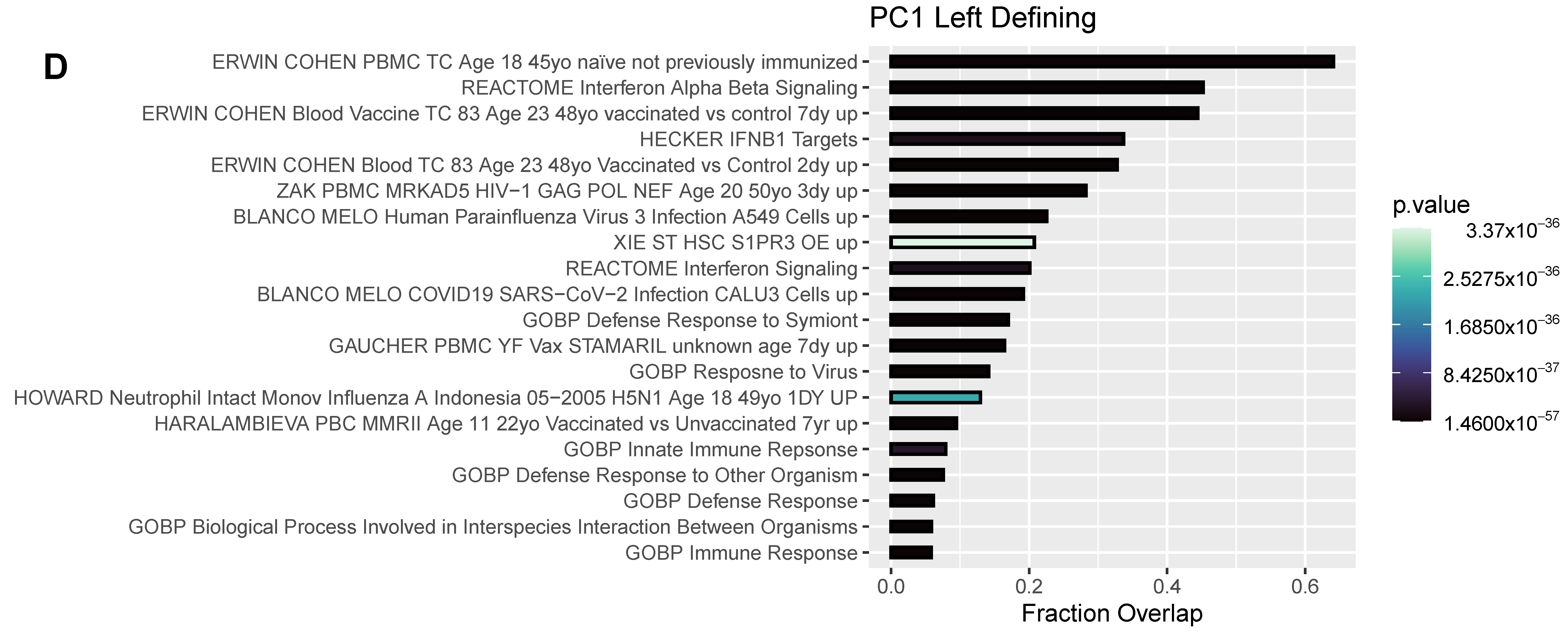
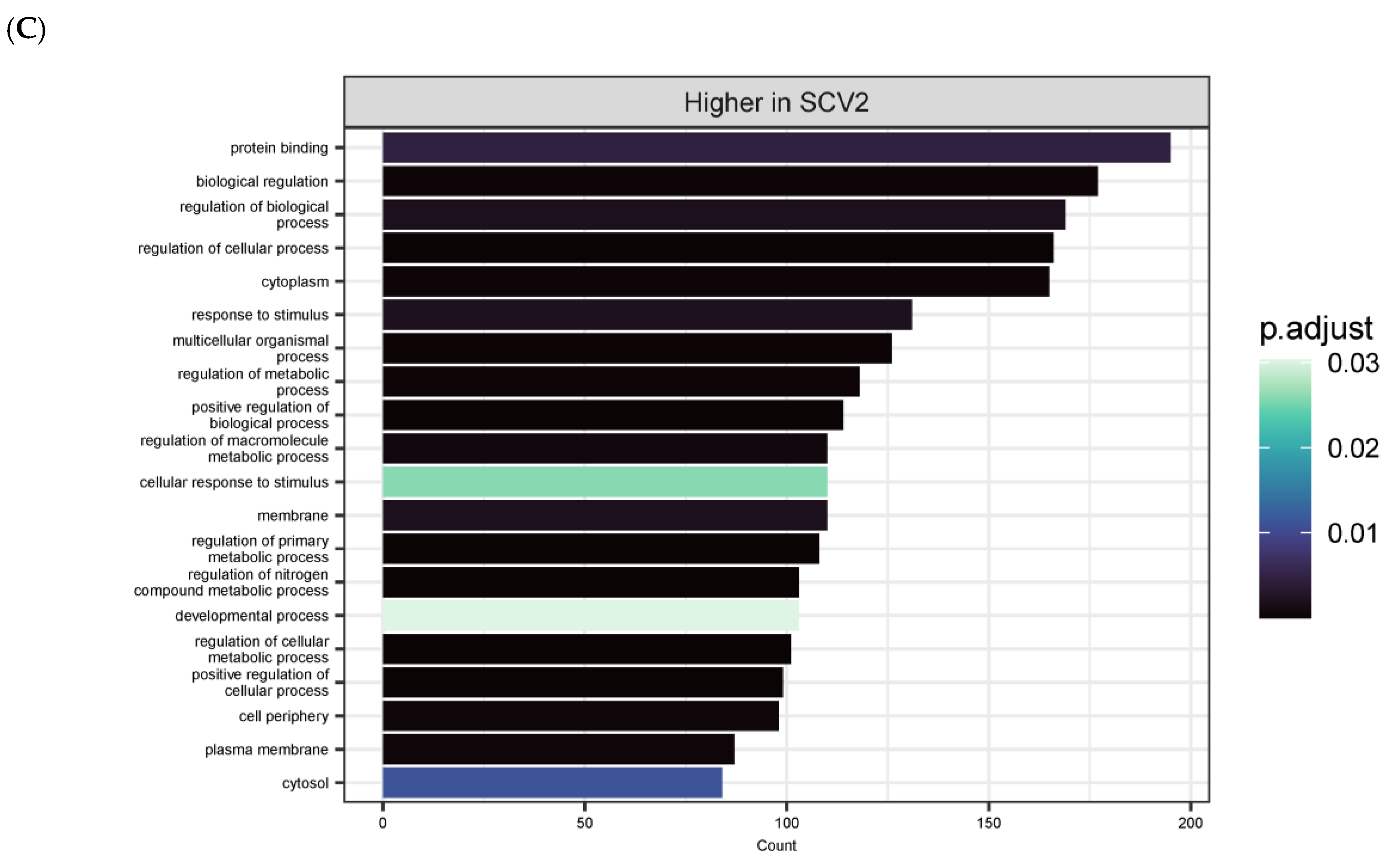
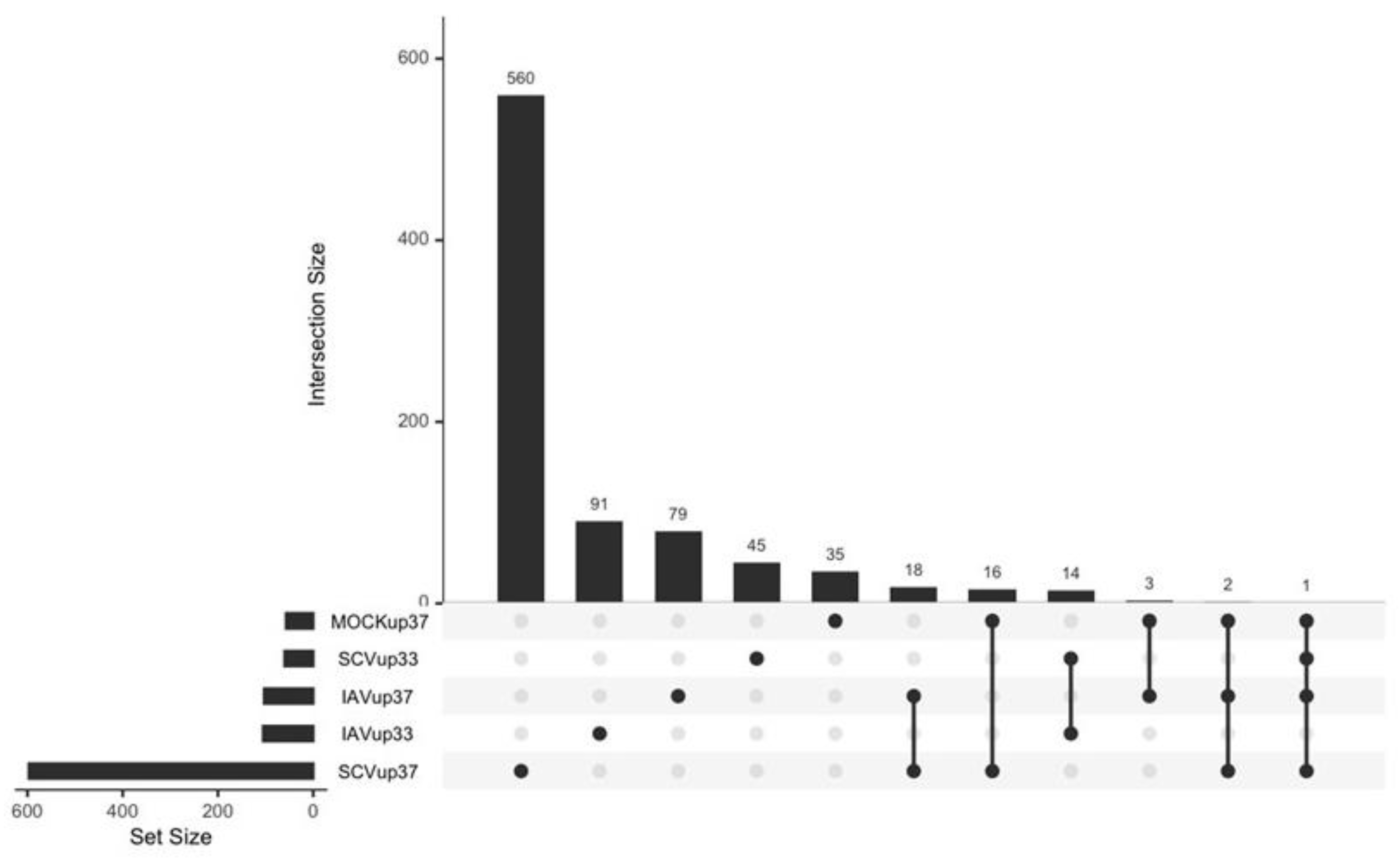
3.4. Differentially Expressed Genes (DEGs) and Pathway Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noor, R.; Maniha, S.M. A brief outline of respiratory viral disease outbreaks: 1889-till date on the public health perspectives. Virus Dis. 2020, 31, 441–449. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. S4), D49–D53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokars, J.I.; Olsen, S. Carrie Reed, Seasonal Incidence of Symptomatic Influenza in the United States. Clin. Infect. Dis. 2018, 66, 1511–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.cdc.gov/flu/spotlights/2022-2023/early-flu-activity.htm#:~:text=October%2014%2C%202022%E2%80%94CDC’s%20first,parts%20of%20the%20United%20States (accessed on 30 December 2022).
- Gounder, A.P.; Boon, A.C.M. Influenza pathogenesis: The effect of host factors on severity of disease. J. Immunol. 2019, 202, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; et al.; China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Du, H.; Gardner, L. COVID-19 Dashboard. 2020. Available online: https://coronavirus.jhu.edu/map.html (accessed on 30 December 2022).
- Matthay, M.A.; Aldrich, J.M.; Gotts, J.E. Treatment for severe acute respiratory distress syndrome from COVID-19. Lancet Respir. Med. 2020, 8, 433–434. [Google Scholar] [CrossRef] [Green Version]
- Manzanares-Meza, L.D.; Medina-Contreras, O. SARS-CoV-2 and influenza: A comparative overview and treatment implications. SARS-CoV-2 e influenza: Revisión comparativa e implicaciones del tratamiento. Bol. Med. Hosp. Infant. Mex. 2020, 77, 262–273. [Google Scholar] [CrossRef]
- Kaeuffer, C.; Le Hyaric, C.; Fabacher, T.; Mootien, J.; Dervieux, B.; Ruch, Y.; Hugerot, A.; Zhu, Y.J.; Pointurier, V.; Clere-Jehl, R.; et al. Clinical characteristics and risk factors associated with severe COVID-19: Prospective analysis of 1045 hospitalised cases in North-Eastern France, March 2020. Euro. Surveill. 2020, 25, 2000895. [Google Scholar] [CrossRef]
- Zsichla, L.; Müller, V. Risk Factors of Severe COVID-19: A Review of Host, Viral and Environmental Factors. Viruses 2023, 15, 175. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Rocha, J.; Gonzalez-Colin, C.; Bhattacharyya, S.; Madrigal, A.; Ottensmeier, C.H.; Ay, F.; Chandra, V.; Vijayanand, P. COVID-19 genetic risk variants are associated with expression of multiple genes in diverse immune cell types. Nat. Commun. 2021, 12, 6760. [Google Scholar] [CrossRef]
- Dunning, J.; Blankley, S.; Hoang, L.T.; Cox, M.; Graham, C.M.; James, P.L.; Bloom, C.I.; Chaussabel, D.; Banchereau, J.; Brett, S.J.; et al. Progression of whole-blood transcriptional signatures from interferon-induced to neutrophil-associated patterns in severe influenza. Nat. Immunol. 2018, 19, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.C.; Simons, N.W.; Wilkins, L.; Cheng, E.; Del Valle, D.; Hoffman, G.; Cervia, C.; Fennessy, B.; Mouskas, K.; Francoeur, N.; et al. Molecular states during acute COVID-19 reveal distinct etiologies of long-term sequelae. Nat. Med. 2022, 29, 236–246. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Li, X.; Mou, T.; Zhou, X.; Li, D.; Wang, L.; Zhang, Y.; Dong, X.; Zheng, H.; Guo, L.; et al. Distinct infection process of SARS-CoV-2 in human bronchial epithelial cell lines. J. Med. Virol. 2020, 92, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Ryu, G.; Shin, H.W. SARS-CoV-2 Infection of Airway Epithelial Cells. Immun. Netw. 2021, 21, e3. [Google Scholar] [CrossRef]
- Ruiz García, S.; Deprez, M.; Lebrigand, K.; Cavard, A.; Paquet, A.; Arguel, M.J.; Magnone, V.; Truchi, M.; Caballero, I.; Leroy, S.; et al. Novel dynamics of human mucociliary differentiation revealed by single-cell RNA sequencing of nasal epithelial cultures. Development 2019, 146, dev177428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.C.; Karamanska, R.; Busch, M.G.; Dell, A.; Olsen, C.W.; Haslam, S.M. Glycan analysis and influenza A virus infection of primary swine respiratory epithelial cells: The importance of NeuAc{alpha}2-6 glycans. J. Biol. Chem. 2010, 285, 34016–34026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumlin, U.; Olofsson, S.; Dimock, K.; Arnberg, N. Sialic acid tissue distribution and influenza virus tropism. Influenza Other Respir. Viruses 2008, 2, 147–154. [Google Scholar] [CrossRef]
- van Riel, D.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D.; Kuiken, T. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am. J. Pathol. 2007, 171, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Soeren, L.; Chua, R.; Trefzer, T.; Kahn, N.; Schnieder, M.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.; et al. SARS-CoV-2 Receptor ACE2 and TMPRSS2 Are Predominantly Expressed in a Transient Secretory Cell Type in Subsegmental Bronchial Branches. Biorxiv 2020, biorxiv: 2020-03. [Google Scholar] [CrossRef] [Green Version]
- Belshe, R.B.; Mendelman, P.M.; Treanor, J.; King, J.; Gruber, W.C.; Piedra, P.; Bernstein, I.D.; Hayden, F.G.; Kotloff, K.; Zangwill, K.; et al. The Efficacy of Live Attenuated, Cold-Adapted, Trivalent, Intranasal Influenzavirus Vaccine in Children. New Engl. J. Med. 1998, 338, 1405–1412. [Google Scholar] [CrossRef]
- Hao, S.; Ning, K.; Kuz, C.; Vorhies, K.; Yan, Z.; Qiu, J. Long-term modeling of SARS-CoV2 infection of in vitro cultured polarized human airway epithelium. mBio 2020, 11, e02852-20. [Google Scholar] [CrossRef] [PubMed]
- Taye, B.; Yeo, D.; Lee, R.T.C.; Tan, B.H.; Sugrue, R.J.; Maurer-Stroh, S. Inter-Species Host Gene Expression Differences in Response to Human and Avian Influenza A Virus Strains. Int. J. Mol. Sci. 2017, 18, 2295. [Google Scholar] [CrossRef] [Green Version]
- Pociask, D.A.; Scheller, E.V.; Mandalapu, S.; McHugh, K.J.; Enelow, R.I.; Fattman, C.L.; Kolls, J.K.; Alcorn, J.F. IL-22 is essential for lung epithelial repair following influenza infection. Am. J. Pathol. 2013, 182, 1286–1296. [Google Scholar] [CrossRef] [Green Version]
- Gamage, A.M.; Tan, K.S.; Chan WO, Y.; Liu, J.; Tan, C.W.; Ong, Y.K.; Thong, M.; Andiappan, A.K.; Anderson, D.E.; Wang, Y.; et al. Infection of human Nasal Epithelial Cells with SARS-CoV-2 and a 382-nt deletion isolate lacking ORF8 reveals similar viral kinetics and host transcriptional profiles. PLoS Pathog. 2020, 16, e1009130. [Google Scholar] [CrossRef] [PubMed]
- Stölting, H.; Baillon, L.; Frise, R.; Bonner, K.; Hewitt, R.J.; Molyneaux, P.L.; Gore, M.L.; Barclay, W.; Saglani, S.; et al.; Breathing Together Consortium Distinct airway epithelial immune responses after infection with SARS-CoV-2 compared to H1N1. Mucosal Immunol. 2022, 15, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Keck, T.; Leiacker, R.; Riechelmann, H.; Rettinger, G. Temperature profile in the nasal cavity. Laryngoscope 2000, 110, 651–654. [Google Scholar] [CrossRef]
- Evangelica, T.; Vasileios, K.; Richard, L. Temperature and humidity measurements in nasal cavity. In Proceedings of the MeMeA-2009-International Workshop on Medical Measurements and Applications, Cetraro, Italy, 29–30 May 2009. [Google Scholar]
- Foxman, E.F.; Storer, J.A.; Fitzgerald, M.E.; Wasik, B.R.; Hou, L.; Zhao, H.; Turner, P.E.; Pyle, A.M.; Iwasaki, A. Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells. Proc. Natl. Acad. Sci. USA 2015, 112, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, N.G.; Sanderson, G.; Hunter, J.; Johnston, S.L. Rhinoviruses Replicate Effectively at Lower Airway Temperatures. J. Med. Virol. 1999, 58, 100–104. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, I.I.W.A.; King, L.S.; Lane, A.P.; Pekosz, A. Restricted replication of the live attenuated influenza A virus vaccine during infection of primary differentiated human nasal epithelial cells. Vaccine 2015, 33, 4495–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.; Zhou, R.; Liu, H.; Rothman, R.; Fenstermacher, K.; Pekosz, A. Antigenic alteration of 2017-2018 season influenza B vaccine by egg-culture adaption. Front. Virol. [CrossRef]
- Gniazdowski, V.; Paul Morris, C.; Wohl, S.; Mehoke, T.; Ramakrishnan, S.; Thielen, P.; Powell, H.; Smith, B.; Armstrong, D.T.; Herrera, M.; et al. Repeated Coronavirus Disease 2019 Molecular Testing: Correlation of Severe Acute Respiratory Syndrome Coronavirus 2 Culture With Molecular Assays and Cycle Thresholds. Clin. Infect. Dis. 2021, 73, e860–e869. [Google Scholar] [CrossRef]
- Ursin, R.L.; Liu, H.; Powell, H.R.; Westerbeck, J.W.; Shaw-Saliba, K.; Sylvia, K.E.; Fenstermacher, K.J.; Mehoke, T.; Thielen, P.; Rothman, R.E.; et al. Differential Antibody Recognition of H3N2 Vaccine and Seasonal Influenza Virus Strains Based on Age, Vaccine Status, and Sex in the 2017-2018 Season. J. Infect. Dis. 2020, 222, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A Simple Method of Estimating Fifty Per Cent Endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- Wohlgemuth, N.; Ye, Y.; Fenstermacher, K.J.; Liu, H.; Lane, A.P.; Pekosz, A. The M2 protein of live, attenuated influenza vaccine encodes a mutation that reduces replication in human nasal epithelial cells. Vaccine 2017, 35 (Pt B), 6691–6699. [Google Scholar] [CrossRef]
- Canaday, L.M.; Resnick, J.D.; Liu, H.; Powell, H.; McCoy, A.M.; Nguyen, D.; Pekosz, A. HA and M2 sequences alter the replication of 2013-16 H1 live attenuated influenza vaccine infection in human nasal epithelial cell cultures. Vaccine 2022, 40, 4544–4553. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 30 December 2022).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 December 2022).
- Available online: https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers/GRCh38_latest_genomic.fna.gz (accessed on 30 December 2022).
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzburg, S. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Squires, R.B.; Noronha, J.; Hunt, V.; García-Sastre, A.; Macken, C.; Baumgarth, N.; Suarez, D.; Pickett, B.E.; Zhang, Y.; Larsen, C.N.; et al. Influenza research database: An integrated bioinformatics resource for influenza research and surveillance. Influenza. Respir. Viruses 2012, 6, 404–416. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.; Whitwham, A.; Keane, T.; McCarthy, S.; Davies, R.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.; Han, Y.; He, Q. Clusterprofiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. Gprofiler2–An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, ELIXIR–709. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.; Lex, A.; UpSetR, N.G. An R Package for the Visualization of Intersecting Sets and their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 30 December 2022).
- David, G.; Lisa, B.; Daniela, S.; Andrew, P. A Targeted siRNA Screen to Identify SNAREs Required for Constitutive Secretion in Mammalian Cells. Traffic (Cph. Den.) 2010, 11, 1191–1204. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulasinghe, A.; Tan, C.W.; Miggiolaro, A.F.R.S.; Monkman, J.; SadeghiRad, H.; Bhuva, D.D.; da Silva Motta Junior, J.; de Paula, C.B.V.; Nagashima, S.; Baena, C.P.; et al. Profiling of lung SARS-CoV-2 and influenza virus infection dissects virus-specific host responses and gene signatures. Eur. Respir. J. 2022, 59, 2101881. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.; Chen, J.; Thapar, V.; Nieman, L.; Solovyov, A.; Mehta, A.; Lieb, D.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Fisher, J.M.; Dang, H.X.; White, N.M.; Strand, M.; Krasnick, B.; Rozycki, E.; Jeffers, G.; Grossman, J.; Highkin, M.; Tang, C. Non-coding RNA RAMS11 promotes metastatic colorectal cancer progression. Nat. Commun. 2020, 11, 2156. [Google Scholar] [CrossRef]
- Khosravi, M.; Lin, R.-L.; Maskey, A.; Pandey, S.; Lin, A.-H.; Lee, L.-Y. A distinct difference between air and mucosal temperatures in human respiratory tract. Front. Med. 2021, 8, 650637. [Google Scholar] [CrossRef]
- Presland, R.B.; Dale, B.A. Epithelial structural proteins of the skin and oral cavity: Function in health and disease. Crit. Rev. Oral Biol. Med. 2000, 11, 383–408. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Gokul, S. Keratinization and its disorders. Oman Med. J. 2012, 27, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Deo, P.N.; Deshmukh, R. Pathophysiology of keratinization. J. Oral. Maxillofac. Pathol. 2018, 22, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Pozuelos, G.L.; Kagda, M.; Rubin, M.A.; Goniewicz, M.L.; Girke, T.; Talbot, P. Transcriptomic Evidence That Switching from Tobacco to Electronic Cigarettes Does Not Reverse Damage to the Respiratory Epithelium. Toxics 2022, 10, 370. [Google Scholar] [CrossRef]
- Morrison, C.B.; Edwards, C.E.; Shaffer, K.M.; Araba, K.C.; Wykoff, J.A.; Williams, D.R.; Asakura, T.; Dang, H.; Morton, L.C.; Gilmore, R.C.; et al. SARS-CoV-2 infection of airway cells causes intense viral and cell shedding, two spreading mechanisms affected by IL-13. Proc. Natl. Acad. Sci. USA 2022, 119, e2119680119. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, U.; Martinelli, R.; Vollmann, E.H.; Best, K.; Therien, A.G. The impact of DAMP-mediated inflammation in severe COVID-19 and related disorders. Biochem. Pharmacol. 2022, 195, 114847. [Google Scholar] [CrossRef]
- Cheemarla, N.R.; Watkins, T.A.; Mihaylova, V.T.; Wang, B.; Zhao, D.; Wang, G.; Landry, M.L.; Foxman, E.F. Dynamic innate immune response determines susceptibility to SARS-CoV-2 infection and early replication kinetics. J. Exp. Med. 2021, 218, e20210583. [Google Scholar] [CrossRef]
- Wu, C.T.; Lidsky, P.V.; Xiao, Y.; Cheng, R.; Lee, I.T.; Nakayama, T.; Jiang, S.; He, W.; Demeter, J.; Knight, M.G.; et al. SARS-CoV-2 replication in airway epithelia requires motile cilia and microvillar reprogramming. Cell 2023, 186, 112–130.e20. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Sawyer, S.L. How host genetics dictates successful viral zoonosis. PLoS Biol. 2019, 17, e3000217. [Google Scholar] [CrossRef] [PubMed]
- Stauft, C.B.; Sangare, K.; Wang, T.T. Differences in New Variant of Concern Replication at Physiological Temperatures In Vitro. J. Infect. Dis. 2023, 227, 202–205. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Gultom, M.; Kelly, J.N.; Steiner, S.; Russeil, J.; Mangeat, B.; Cora, E.; Pezoldt, J.; Holwerda, M.; Kratzel, A.; et al. Disparate temperature-dependent virus-host dynamics for SARS-CoV-2 and SARS-CoV in the human respiratory epithelium. PLoS Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nori, W.; Ghani Zghair, M.A. Omicron targets upper airways in pediatrics, elderly and unvaccinated population. World J. Clin. Cases 2022, 10, 12062–12065. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine. 2021, 65, 103255. [Google Scholar] [CrossRef]
- Breining, P.; Frølund, A.L.; Højen, J.F.; Gunst, J.D.; Staerke, N.B.; Saedder, E.; Cases-Thomas, M.; Little, P.; Nielsen, L.P.; Søgaard, O.S.; et al. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin. Pharmacol. Toxicol. 2021, 128, 204–212. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Gary, R. Whittaker, Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Nakagawara, K.; Chubachi, S.; Namkoong, H.; Tanaka, H.; Lee, H.; Azekawa, S.; Otake, S.; Fukushima, T.; Morita, A.; Wa-tase, M.; et al. Impact of upper and lower respiratory symptoms on COVID-19 outcomes: A multicenter retrospective cohort study. Respir. Res. 2022. [Google Scholar] [CrossRef]
- Rashid, F.; Xie, Z.; Suleman, M.; Shah, A.; Khan, S.; Luo, S. Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front. Immunol. 2022, 13, 940756. [Google Scholar] [CrossRef]
- Herder, V.; Dee, K.; Wojtus, J.K.; Goldfarb, D.; Rozario, C.; Gu, Q.; Jarrett, R.F.; Epifano, I.; Stevenson, A.; McFarlane, S.; et al. Elevated temperature inhibits SARS-CoV-2 replication in respiratory epithelium independently of the induction of IFN-mediated innate immune defences. bioRxiv 2020, bioRxiv: 2020.12. 04.411389. [Google Scholar] [CrossRef]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resnick, J.D.; Beer, M.A.; Pekosz, A. Early Transcriptional Responses of Human Nasal Epithelial Cells to Infection with Influenza A and SARS-CoV-2 Virus Differ and Are Influenced by Physiological Temperature. Pathogens 2023, 12, 480. https://doi.org/10.3390/pathogens12030480
Resnick JD, Beer MA, Pekosz A. Early Transcriptional Responses of Human Nasal Epithelial Cells to Infection with Influenza A and SARS-CoV-2 Virus Differ and Are Influenced by Physiological Temperature. Pathogens. 2023; 12(3):480. https://doi.org/10.3390/pathogens12030480
Chicago/Turabian StyleResnick, Jessica D., Michael A. Beer, and Andrew Pekosz. 2023. "Early Transcriptional Responses of Human Nasal Epithelial Cells to Infection with Influenza A and SARS-CoV-2 Virus Differ and Are Influenced by Physiological Temperature" Pathogens 12, no. 3: 480. https://doi.org/10.3390/pathogens12030480
APA StyleResnick, J. D., Beer, M. A., & Pekosz, A. (2023). Early Transcriptional Responses of Human Nasal Epithelial Cells to Infection with Influenza A and SARS-CoV-2 Virus Differ and Are Influenced by Physiological Temperature. Pathogens, 12(3), 480. https://doi.org/10.3390/pathogens12030480