Abstract
Kala-azar, also known as visceral leishmaniasis (VL), is a disease caused by Leishmania infantum and L. donovani. Patients experience symptoms such as fever, weight loss, paleness, and enlarged liver and spleen. The disease also affects immunosuppressed individuals and has an overall mortality rate of up to 10%. This overview explores the literature on the pathogenesis of preclinical and clinical stages, including studies in vitro and in animal models, as well as complications and death. Asymptomatic infection can result in long-lasting immunity. VL develops in a minority of infected individuals when parasites overcome host defenses and multiply in tissues such as the spleen, liver, and bone marrow. Hepatosplenomegaly occurs due to hyperplasia, resulting from parasite proliferation. A systemic inflammation mediated by cytokines develops, triggering acute phase reactants from the liver. These cytokines can reach the brain, causing fever, cachexia and vomiting. Similar to sepsis, disseminated intravascular coagulation (DIC) occurs due to tissue factor overexpression. Anemia, hypergammaglobulinemia, and edema result from the acute phase response. A regulatory response and lymphocyte depletion increase the risk of bacterial superinfections, which, combined with DIC, are thought to cause death. Our understanding of VL’s pathogenesis is limited, and further research is needed to elucidate the preclinical events and clinical manifestations in humans.
1. Infection
Kala-azar, or visceral leishmaniasis (VL), is the second most lethal tropical and subtropical disease and seventh in the loss of disability-adjusted life years [1,2]. It is caused by the protozoa Leishmania infantum and L. donovani and is transmitted by the bite of infected sand flies. VL caused by L. infantum is a zoonosis involving a variety of mammals, especially the dog, distributed across Central Asia, the Middle East, the Mediterranean Basin, and South and Central America. The disease caused by L. donovani is restricted to humans in South Asia and East Africa [3]. VL has a protracted course of clinical development with fever, wight loss, paleness, and hepatosplenomegaly. Five to ten percent of patients die of this disease, resulting from the lethal complications of bacterial co-infections and hemorrhages. Anemia, neutropenia, thrombocytopenia, hypoalbuminemia, hyperglobulinemia, high C-reactive protein (CRP), and high erythrocyte sedimentation rate (ESR) are regularly noticed. It is a significant opportunistic infection in immunocompromised individuals, particularly those living with HIV or taking immunosuppressive drugs [4]. While transmission has been rapidly and consistently falling in recent years in South Asia [5,6], the incidence in Brazil has a worrisome trend toward a gradual increase in mortality [7].
This review serves to underscore the importance of elucidating disease mechanisms for the prospect of developing new treatments. The information provided herein is based on a literature search of original publications on VL and other reviews for general subjects. The literature search was focused on articles related to clinical disease. Publications on experimental in vitro and animal infections were consulted where information was not available in human infection, i.e., all the preclinical early events. Information of relevance is included from publications on other Leishmania spp. and other infectious diseases. Finally, the review is centered on immunocompetent human subjects, although a few comments are made about HIV co-infected patients.
The development of VL is more exception than rule after exposure to viscerotropic Leishmania. Indeed, a certain proportion of the human population in a given VL endemic area are leishmanin skin tests positive, indicating previous Leishmania infection, but only a small proportion of them have a history of the disease development [8,9,10,11]. Based on a population-based leishmanin skin-test study, the ratio of asymptomatic to symptomatic infection was estimated to be more than 200:1 [12].
The early preclinical stages of human infection by viscerotropic Leishmania have defied investigation in situ due to the elusive, long and variable lengths of the incubation period. The early events of infection and host response are available from a large body of extensive work on experimental leishmaniasis in vitro and in animal models. They are included for discussion with the caveat that they may or may not reflect human infection in the real world. Notably, infection of mice or dogs with L. donovani or L. infantum does not reproduce all the clinical symptoms and signs of human visceral leishmaniasis.
In animal models, infected sand flies have been found to inoculate infective promastigotes of various Leishmania spp., e.g., L. infantum, L. donovani, and L. amazonensis into the skin. Most of them are killed in a few minutes by complement-mediated lysis, leaving few survivors phagocytosed by neutrophils or monocytes [13]. The cutaneous species, L. major, encounter the phagocytes via their pathogen recognition receptors (PRR) when successfully infecting C57BL/6 mice [14,15,16]. It is not known if the infected neutrophils may present antigens directly to T cells, as reported for viral antigens in human neutrophils in vitro [17] or if they are phagocytized by neutrophils that undergo apoptosis and then are ingested by monocytes—the Trojan horse hypothesis [18]. L. major is also directly phagocytized by human monocytes, consistent with the leishmanial intracellular parasitism of mononuclear phagocytes known for successful infection, as demonstrated in humanized transgenic mice [19]. After phagocytosis, Leishmania antigens are presented to B- and T- cells, and adaptative immunity develops [20]. The interaction of Leishmania with dendritic cells (DC) has been reviewed [21] in the context of their significance as the antigen-presenting cells in T cell immunity [22]. Leishmania–DC interactions in relation to the lasting immunity after the curing of leishmaniasis is of great interest for further investigation.
The migratory pathway of L. infantum and L. donovani infection is expected to begin from the skin via the regional lymph nodes to the final destinations of spleen, bone marrow, and liver. A struggle is predicted between permissive versus protective innate and adaptative immune response to determine susceptibility or resistance during this Leishmania migration. The battle begins involving the armamentarium, composed of the parasite’s molecules, such as lipophosphoglycan, GP63, and others, and the host’s molecules initially confronting the parasite: Toll-like receptors (TLRs) (TLR2, TLR4, and TLR9) and other types of receptors, together with the antimicrobial hydrolases/peptides of infected macrophages, reactive oxygen species, and nitric oxide produced by these and other mononuclear innate immune cells. Disease ensues when parasite molecules subvert the host response, turning it amenable for Leishmania intracellular survival via several levels of molecular interactions, e.g., [A] induction of regulatory cytokine IL-10 by parasitized macrophages; [B] inhibition of Th1-acquired immune response and IFN-γ release; and [C] promotion of Th2 T cells and regulatory T cell (Tregs) clonal expansion by further secretion of IL-10, IL-4, and TGF-β regulatory cytokines. This process finally results in T cell anergy and leads to disease development. Infected neutrophils and monocytes/macrophages were observed in the patients’ blood, presumably serving to spread amastigotes to other infection sites and facilitating transmission to other hosts as female sand flies take bloodmeals [23]. The host may win the battle against the infection when macrophages that phagocytose Leishmania produce IL-12, thereby stimulating the expansion of proinflammatory Th1 T cell clones for IFN-γ release, gaining the ability to kill the parasites. After this point, cell-mediated immunity is expected to be firmly established to control parasite proliferation. The host–parasite struggle for viscerotropic Leishmania to establish parasitism in the mammal hosts has been elegantly reviewed by previous workers [24,25,26,27].
Disease development is modulated by the parasite–vector–mammalian host interactions. Genomic studies of viscerotropic versus cutanotropic Leishmania delineate the significance of the products of the A2 gene family in parasite visceralization. A mutation in the ras-like Rag C GTPase in the mTOR pathway of BALB7c mice may contribute to the control of Leishmania visceralization [28]. These observations offer clues for investigation to explain why L. donovani variants cause human cutaneous leishmaniasis instead of VL in Sri Lanka [29]. Finally, whole-genome sequence analyses of 109 L. infantum isolates found parasite sequences that are strongly associated with the phenotypes of disease severity, such as mortality and kidney failure [30]. However, observations on the early preclinical events in human infection will be needed to fully understand the important question of how the disease is initiated. This might eventually be accomplished through the use of controlled human infection models (CHIM), as performed with SARS-CoV-2 [31] notwithstanding the limitations of this approach. The development of more sophisticated human 3D organoid culture systems [32] may be useful to study infection, if vasculature can be introduced for the influx of immune cells necessary for studying infection and immunity. Another useful approach under development is the digital microfluidic sensing to monitor in real time inflammation and over-reactive immune response [33].
Evidence indicates that human hosts contribute to the development of VL. Several studies have demonstrated that the human genetic background plays a role in the fate of the established infection by modulating its development into a disease state [34,35,36,37]. There is evidence of age-dependent susceptibility to VL, as indicated by the higher proportion of disease in the youngest in a given population [9,10,38]. Patients at the extreme ends of the age groups also tend to have higher parasite burdens, presumably due to their weaker immunity [39]. Both disease incidence and parasite loads are higher in males at reproductive age than in the other demographic groups [39,40,41,42,43], leading to the proposal that male sex hormones, including testosterone and dihydrotestosterone, may play a role in fueling the infection [44]. Another important host factor influencing the development of disease is immunity to prior infection. Solid immunity is clearly generated by previous Leishmania infections, regardless of symptomatic or asymptomatic individuals in an immunocompetent population, as both reactivation and reinfection are rarely seen [45,46]. On the same track, acquired immunosuppression strongly influences evolution of the infection in this population from asymptomatic to symptomatic [47]. Finally, malnutrition has long been considered a risk factor for neglected tropical diseases, although its role in human VL is unclear [9,10,38].
Sand flies can also modulate the outcome of Leishmania infection when delivering infective promastigotes experimentally into the skin of mice with diverse species of Leishmania. Via the bites of sand flies, it has been shown that their saliva is rich in pharmacologically active substances, which modulate vasodilation, anticoagulation, and immune response in favor of Leishmania successful infection. Sand fly gut microbiota may also play a certain role [48]. The success or failure of Leishmania infection in mice is ultimately determined by the dose of infective parasites delivered by the vectors [49,50]. Figure 1 shows the likely evolution of VL from the sand fly bite to disease development.
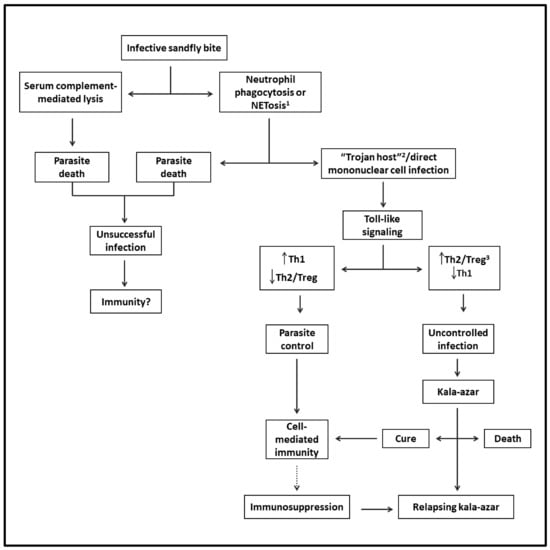
Figure 1.
Flowchart depicting the likely evolution of VL from infection to disease: After the infective sand fly bite, parasites are released into the skin. Some perish and thus are not expected to generate a cellular immune response. Viable parasites reach their host cells directly via phagocytosis by mononuclear phagocytes and/or via phagocytosis by neutrophils (Trojan host), followed by engulfment by mononuclear cells. After triggering Toll-like intracellular signaling in either case, the infection is controlled by Th1- and cell-mediated immunity. However, the parasites may overcome this host response to replicate by mediating the generation of Th2 and Treg responses, progressing to VL. The patients may die or be cured, leading to lasting immunity to reinfection. Some patients may suffer from relapsing VL due to suppression of cell-mediated immunity preventing a complete cure. 1 Regulated cell death with the formation of neutrophil extracellular traps (NET). 2 A mechanism of intracellular cell entry through the phagocytosis of neutrophils with engulfed microbe by mononuclear phagocytes. 3 T-regulatory cells.
2. Disease
Successful infection leads to unchecked proliferation of amastigotes in the mononuclear phagocytes, accounting for the disease state of VL. That is marked by systemic innate response with an increase in the plasma level of proinflammatory cytokines and the regulatory cytokine IL-10 [51,52]. Parasites must overcome host resistance in order to proliferate for the disease to ensue. The emergence of Th2 and Treg susceptible phenotype over Leishmania-specific acquired cellular immunity is expected, as indicated by the failure of immune T cells to proliferate and to produce IFN-γ in clinical cases [53,54,55,56,57]. Since Leishmania produce no toxins to account for the pathology of the disease seen, the innate host immune response is solely responsible for the clinical symptoms observed [58]. Indeed, there is a positive correlation between severity of the disease and parasite burdens [39,59]. Figure 2 shows some examples of children with various signs and symptoms of VL.
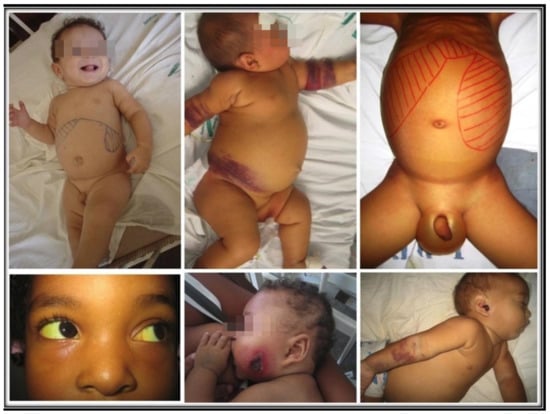
Figure 2.
Uncomplicated, complicated, and lethal VL. Top left: an uncomplicated disease with hepatosplenomegaly and paleness. Top center: extensive bruising. Top right: large hepatosplenomegaly, with ascites and edema of the scrotum. Bottom left: scleral jaundice. Bottom center and right: Pseudomonas aeruginosa secondary infection in the face and the ear.
3. Constitutional Symptoms
Table 1 summarizes the hypothetical pathogenesis of VL. VL’s signs and symptoms overlap with those of overproducing proinflammatory cytokines, like IL-6, IL-1β, TNF-α, IL-8, and other proinflammatory cytokines as well as the regulatory cytokines, IL-10 and TGF-β. All these cytokines are produced via signal transduction triggered by Leishmania interaction with PRRs after infection of macrophages [60]. IL-6 is thought to have the broadest spectrum of impact due to the nature of its receptors. IL-6 interacts with both membrane-bound (mIL-6R) and soluble IL-6 receptors, and the co-receptor gp130, accounting for its pleiotropic activities [61]. The binding of IL-6 to mIL-6R and gp130 induces the synthesis of the acute phase reactants (APR) in hepatocytes [62] and also the overexpression of tissue factors in endothelial cells and monocytes, triggering the coagulation cascade [63,64,65]. The gp130 co-receptor of IL-6 is expressed on the plasma membrane of most cell types, including the circumventricular organs, the preoptic area of the hypothalamus and muscle cells [66,67]. Overproduction of the aforementioned cytokines, especially IL-6 and APR are the cornerstones for the development of the symptoms observed in VL.

Table 1.
Pathogenic mechanisms proposed to account for the clinical manifestations of VL deduced from organ/tissues and potential mediators involved in human patients and animal models.
The APR overexpressed by the hepatocytes include CRP (C-reactive protein), hepcidin, procalcitonin, CD14, serum amyloid A protein, fibrinogen, ferritin, and complement proteins C3 and C4 [68,69,70]. The different APRs produced vary in level. CRP is highly correlated with IL-6 [71] and can increase by a few thousand folds in VL, caused by both L. donovani and L. infantum [72,73,74,75]. Albumin decreases in level during the acute phase response, hence a negative APR, regularly notable in the sera of VL patients [58,76]. ESR (erythrocyte sedimentation rate) is almost always present at a high value in VL. This is a nonprotein APR whose level is known to change in response to plasma fibrinogen levels and plasma viscosity, hence an “indirect” APR [77,78,79]. APR rises with the increase in procoagulant and the decrease in anticoagulant proteins, fueling the already activated coagulation cascade, leading to fibrinolysis and bleeding [80].
Fever is one of the most notable symptoms of VL. It is mediated by the proinflammatory cytokines, called endogenous pyrogens, mostly IL-1β, TNF-α and IL-6. Endogenous pyrogens are synthesized and released after PRR (likely Toll-like receptor 2) interaction with Leishmania in macrophages [60,81]. After reaching the hypothalamus via the circumventricular organs that lack the blood–brain barrier, the pyrogens trigger the release of prostaglandin A2 via binding the prostaglandin E receptor in the preoptic area of the hypothalamus. This is the brain region where fever is generated in neurons within the central thermoregulatory circuitries [82,83].
Weight loss is also a common manifestation of VL, resulting from severe malnutrition [84,85]. Patients with VL-driven malnutrition are at higher risk of death [86,87,88,89,90]. While the mechanisms are unknown, there are at least three main possibilities to account for the weight loss, all linked to persistent systemic inflammation due to the production of proinflammatory cytokines, i.e., (a) loss of appetite as part of the illness following systemic inflammation [67,91,92]; (b) fever that sharply increases the consumption of energy [93]; and (c) acute phase response that increases muscle catabolism leading to sarcopenia, thereby making amino acids abundantly available for APR synthesis in the liver [94,95,96]. The actions of TNF-α, IL-1β, and especially IL-6 on the hypothalamus and muscles appear to play decisive roles in the development of cachexia.
Nausea and emesis are infrequently encountered symptoms of VL with multiple potential causes, including iatrogenic, toxic and infectious causes, gastrointestinal disorders, and central nervous system or psychiatric dysfunctions [97]. Signals to cause these symptoms may be transmitted from the infected viscera with the blood flow to the area postrema, one of the circumventricular organs where the blood–brain barrier is absent and also by the abdominal vagal afferents to the nucleus tractus solitarius. Stimulated neurons then send signals to a central-pattern generator, which coordinates the act of emesis and to the ventral medulla and hypothalamus, reaching the cortical brain areas [98]. Vomiting has been reported to be associated with increased mortality of VL patients in both Africa and Brazil [58,86,88,89,90,99,100,101,102,103]. In these patients, it is associated with higher concentrations of IL-6 according to uni- and multi-variate analyses [104]. The chemical mediators of nausea and vomiting in VL are unknown. These are the alarm symptoms in dengue, which is, like VL, also associated with cytokine storm, involving IL6 and other proinflammatory cytokines [105]. Whether they are the mediators of nausea and vomiting awaits further investigation.
Anemia is also a common clinical manifestation and a risk factor for poor outcome of VL [58,86,88,89,90,100,101,102,103,106,107,108,109,110,111,112]. It is typically due to iron deficiency, with hypochromia and microcytosis marked by a low serum iron concentration [113]. However, the mechanical destruction of red cells, neutrophils, and platelets alone in the enlarged spleen, e.g., hypersplenism, contributes significantly to anemia, independent of inflammation [114]. Moreover, although most VL patients suffer from iron-deficient anemia [113], it is rarely caused by bleeding, indicative of the participation of a different mechanism in VL. This process may involve the APR of VL, hepcidin—an iron-regulating peptide. It functions to inhibit ferroportin, which mobilizes iron from all the iron-transporting cells (enterocytes, spleen, and liver macrophages) into plasma for transport into bone marrow for hematopoiesis [115,116]. Indeed, anemia is negatively correlated with the level of IL-6, the cytokine that regulates APR, including hepcidin [104]; hepcidin mRNA is negatively correlated with hemoglobin in patients with VL [117]. In summary, while the destruction of red cells by hypersplenism is partially responsible for the anemia in VL, the APR contributes to this via the iron-depleting action of hepcidin under the control of proinflammatory cytokines, especially IL-6.
4. Localized Symptoms
Hepatosplenomegaly is one of the most important clues for diagnosing VL. It is determined by lymphoid hyperplasia, as histopathological studies have revealed that the spleen and the liver are full of parasites. There is no evidence of passive hypertensive congestion to explain splenomegaly. In spite of a marked atrophy of the splenic white pulp associated with necrosis and fibrosis of thymus-dependent areas, there is an accumulation of parasites-containing macrophages and plasma cell hyperplasia, accounting for the increase in the spleen volume. The number of parasites in the spleen and its architecture disruption is associated with the activities of proinflammatory and regulatory cytokines produced in diseased humans and dogs [118,119,120,121]. In both cases, parasite proliferation is promoted by the production of TGF-beta together with IL-10, produced locally by CD25(+)Foxp3(−) T cells and systematically by CD25(+)Foxp3(+) T cells [55,122,123]. The spleen increases in size to >10 times of its normal volume and appears to play a central role in the pathogenesis of VL. Splenectomy dramatically improves patients’ clinical conditions, although it does not cure the diseased HIV-co-infected patients [124]. Some degree of hypertrophy is also seen in the liver due to the proliferation of Kupffer cells in patients infected with either L. donovani or L. infantum [119,125].
Protracted edema (swelling) with or without anasarca (generalized edema) is a consequence of APR [109] and a risk factor for death in VL [58,87,88,89,90,101,102,103,107,108,109,111,126,127]. It is caused by a reduced concentration of albumin in plasma. Albumin is a negative APR and the principal protein determinant of oncotic plasma pressure. When it is low, plasma leaks to the interstitial space, leading to edema [128]. Therefore, hypoalbuminemia is VL’s primary cause of edema [68,69,70], consistent with our observation that IL-6 is elevated in edematous patients [104]. Hepatic failure is another cause of hypoalbuminemia, but this is rare in VL [76,119].
Cough is a common symptom and, together with the presence of pulmonary rales and dyspnea, indicates lung involvement in VL. This symptom is caused by a combination of alveolar bacterial pneumonia and interstitial proinflammatory response due to systemic Leishmania infection. These symptoms deserve prompt attention, since they are significantly associated with patients’ death, as found in many independent studies [58,89,101,102,103,109,110,111,126,129]. Interstitial pneumonitis is of inflammatory nature, as suggested by histopathologic studies that have demonstrated interstitial lung involvement where bacterial was absent; amastigotes were scarce, but Leishmania antigens were detected [130]. This conclusion is reinforced by an immunohistochemistry study of autopsy samples, showing interstitial accumulation of macrophages [131] and by computer tomography findings suggestive of association with respiratory symptoms [132]. The immunohistochemistry study also revealed a pattern of regulatory Th2 cytokines, indicative of an increased likelihood of bacterial superinfections. However, the histopathological and immunological data of VL suggest that the interstitial pulmonary involvement may be included in the category of profibrotic and proinflammatory interstitial lung diseases rather than bacterial infectious diseases [133,134,135].
Renal syndromes and low-grade renal failure were frequently observed in VL, and renal failure has been associated with increased mortality, as reported in some studies [89,109,127,136,137]. Proteinuria also reflects disease severity [58]. However, parasites are seldom seen in the kidneys. Renal involvement is related to interstitial nephritis and distinct glomerular participation, like collapsing segmental and focal glomerular sclerosis, necrotizing segmental and focal glomerular sclerosis, and membranoproliferative lesion. The renal pathogenesis is not well understood and is likely multicausal. Due to the presence of polyclonal gamma globulins and IL-6 at high levels, it was hypothesized that proteinuria leads to proximal tubular injury, associated with glomerular inflammation [138,139]. AA amyloid glomerular deposits without mesangial hyperplasia have also been registered, likely as part of the APR [140]. However, the first suspected cause of interstitial nephritis is drug toxicity in VL, since most antileishmanial drugs are nephrotoxic [137].
Hepatitis is not uncommon in VL. The liver enzymes, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are slightly or moderately elevated, indicative of hepatocyte damage and useful as markers for disease severity [58,87,89,90,99,102,106,107,108,109,110,111,126,141,142]. Jaundice or an increased level of bilirubin is one of the most significant risk factors for death, particularly in patients with hemorrhages [12,58,87,89,90,99,101,102,103,107,108,109,110,111,127,129]. Many histopathology studies have demonstrated liver involvement, with ballooning degeneration of hepatocytes, Kupffer cells parasitized with amastigotes, chronic mononuclear infiltrate in the portal space, and fibrosis associated with Ito’s cells transformation into fibroblasts [119,125]. The association between liver inflammation with the elevation of IL-1β, TNF-α and IL-6 has been known in viral hepatitis, suggestive of a similar pathway to explain the liver involvement in VL [62,143]. However, liver involvement usually is not severe, as demonstrated by the rarity of hepatic encephalopathy. Our observations found that IL-6 was the serum cytokine with the strongest correlation, with an elevation of AST but not with that of ALT in a multivariate linear regression analysis [104]. As ALT is more specific to liver involvement than AST, the stronger association of AST with VL severity may reveal a more generalized, multiorgan involvement [144]. Indeed, multiorgan microthrombosis caused by disseminated intravascular coagulation (DIC), as seen in liver biopsies [125], may be another explanation for the rise of AST in VL.
Diarrhea is a prevalent symptom of VL caused by infection with L. infantum, and parasites are seen in the intestinal mucosa [126,145]. It is a consistent risk factor for death [58,86,87,88,89,90,99,100,101,102,103,106,107,108,109,111,127,146]. Severe malnutrition can lead to diarrhea through several mechanisms [147], potentially contributing to diarrhea seen in malnourished patients of VL. Since there is heavy parasitism with mucosal changes in patients with diarrhea and VL, it looks very likely that gut parasitism and local secondary inflammation are the leading cause of diarrhea in VL [126,148]. Comparison of gut mucosa of VL patients with and without diarrhea may further clarify the mechanism of its pathogenesis.
Post-kala-azar dermal leishmaniasis (PKDL) occurs in immunocompetent patients when infected by L. donovani, but only in patients with AIDS when infected by L. infantum [149,150]. This disparity cannot be explained fully without an in-depth study on the complexity of host–parasite–vector interactions, although the genetic differences between the two species is expected to play a role. At a time of a successful VL elimination program in South Asia, PKDL deserves renewed attention, since it is a source of parasites that fuels transmission and threatens elimination efforts. The lesions of South Asia PKDL caused by L. donovani are of the papulonodular and polymorphic type, with high parasite loads. Indian PKDL emerges in 5–10% of the VL patients 2–3 years after chemotherapy. They are rarely cured spontaneously. The East African type is monomorphic macular lesions with low parasite loads and patchy inflammatory infiltrates. East African PKDL emerges in 50–60% of the VL patients after or during the treatment, but 85% of the cases are self-healing. It has been proposed that PKDL occurs after cure of VL with reactivation of the specific immunity, i.e., a decrease in the levels of regulatory T cells, TGF-beta, and IL-10 and an increase in those of IFN-γ, TNF-α, and IL-12. L. donovani persisting in the skin may be detected by reactivated immune cells, which infiltrate into the cutaneous tissue, causing dermal inflammation by the secretion of IFN-γ, giving rise to dermal manifestations [19,151]. However, this conclusion clashes with the recent report that IL-10 remains elevated in PKDL [15]. Of relevance to mention is the report that treatment of VL with liposomal amphotericin B reduces the incidence of PKDL [152]
5. Laboratory Changes
Pancytopenia (reduced blood erythrocytes, white blood cells, and platelets) is a typical feature of VL. It has been frequently observed in hypersplenism of other etiologies under many noninflammatory conditions, e.g., the presinusoidal portal hypertension caused by portal obstruction [77,114,153]. In VL patients, all cytopenia are more severe due to systemic and splenic inflammation, exacerbating the mechanical destruction of blood cells and platelets [120,121]. The essential role of inflammation is highlighted by the facts that when the inflamed spleen of relapsing VL is taken out by splenectomy [120], leukopenia and thrombocytopenia subside rapidly and completely [124], while in noninflammatory hypersplenism, the recovery is only partial [114]. As discussed above, the anemia seen in VL is also a consequence of inflammation via the APR hepcidin, leading to iron-deficiency anemia. In VL, severe anemia has bone marrow erythroid hyperplasia and dysplasia instead of hypoplasia, as revealed by comparing the bone marrow cell population with the peripheral blood counts. This observation suggests that bone marrow plays no role in the anemia, which results from mechanical destruction of red cells in the spleen due to splenomegaly and inflammation-dependent iron deficiency [77,154].
Neutropenia is a prominent white cell alteration in VL. Severe neutropenia (<500 neutrophils/mm3) alone is not a risk factor for VL, in contrast to febrile neutropenia for bacterial sepsis [58,102,103,106,107,108,109,110,111,146]. Neutrophils are produced from myeloid lineage progenitor cells in bone marrow, which mature into nondividing cells and enter a postmitotic pool for distribution. Mature neutrophils are present as a free-flowing blood pool in the vasculature and as the marginated pool, residing in other tissues/organs, e.g., spleen, liver, and bone marrow. Up to 30% of the total mature neutrophils normally reside in human spleen, which increases proportionally with the degrees of splenomegaly, explaining the peripheral neutropenia in hypersplenism. The degree of neutropenia indeed varies with the spleen size in rats and humans with hypersplenism [114,155]. Marginated pools of neutrophils (spleen, liver, and bone marrow) may increase by inflammatory parasitization of these organs, diminishing peripheral neutrophils in conjunction with their necroptosis, pyroptosis, and neutrophil extracellular traps [156,157]. In VL, the myeloid lineage can be hypo- or hyperplastic, independent of peripheral blood neutrophil counts [77,154]. This set of observations indicates that the neutropenia seen in VL is not determined by bone marrow changes but by hypersplenism and systemic inflammation. This is consistent with the observation that L. donovani infection of mice increases their granulocyte colony-stimulating factor (GSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) [158]. The dynamics of neutrophil changes in the spleen, bone marrow and liver needs detailed investigation in VL patients.
Thrombocytopenia is a common feature of VL but is rarely low to the level that requires platelet transfusion [106,159,160]. This is considered as one of the risk factors for poor prognosis [58,87,89,101,102,103,107,109,110,111,127,129]. Thrombocytopenia may result from impaired production, increased destruction, and/or dilution of platelets [161]. Platelets emerge from the fragmentation of the cytoplasm of megakaryocytes in the bone marrow and circulate in the blood as anucleate particles. There are two main mechanisms associated with thrombocytopenia in VL: dilution of peripheral platelets due to hypersplenism and further depletion by consumption coagulopathy. There is no evidence for a reduced production of platelets in VL patients, judging from their bone marrow megakaryocyte cellular integrity [77,154]. The recovery of platelet count is much faster than the spleen size reduction in convalescent patients, suggesting that the primary cause of thrombocytopenia is associated with inflammation leading to DIC, followed by consumption coagulopathy in VL [162].
Hyperglobulinemia, more precisely, hypergammaglobulinemia, is mediated by proinflammatory cytokines in VL patients [109,163]. The hypergammaglobulinemia is polyclonal, resulting from IL-6-mediated differentiation and expansion of B-cells into antibody-producing plasma cells [61,164], producing electrophoretically heterogeneous proteins, chiefly the gamma globulin bands [165]. The numbers of these plasma cells increase in the bone marrow, gut, lymph nodes, spleen, and liver.
Hypoalbuminemia has been found as a risk factor of VL [58,86,87,88,99,100,101,102,103,106,107,108,109,111,126,127,129,166,167]. Hypoalbuminemia is not directly related to hyperglobulinemia because, while B-cells produce gammaglobulins, albumin is synthesized by the liver cells. However, both are initially linked to the innate response driven by IL-6 [70].
CRP and ESR are components of the APR, as stated before. CRP is a pentameric protein synthesized and secreted by the liver, which is elevated in VL. Patients with normal CRP are unlikely candidates of VL diagnosis, requiring further considerations for other diseases. CRP has no prognostic value for the disease [72,74,75,168]. ESR, readily detectable by a cheap and simple test, is also regularly elevated, and when it has low speed may indicate a poor prognosis for patients with VL [112,169,170].
Haemophagocytic lymphohistiocytosis syndrome (HLS) is an inflammatory hematological condition that overlaps with VL presentation in many aspects. The root cause of HLS can be genetic, iatrogenic, inflammatory, or infectious. Virally induced HLS is thought to result from the IFN-γ amplified loop of sustained antigen presentation to CD8+ T cells for activation. CRP, ESR, and ferritin, another APR, are elevated, responsible for the main manifestations of fever, pancytopenia, liver involvement, and marked inflammation [171]. High triglycerides has also been described in VL, particularly in the more severe disease [172,173]. Phagocytosis of erythrocytes, white cells, platelets, and their precursors gave the name to the syndrome but are not all diagnostic requirements. The HLS syndrome of VL does not increase its severity and requires no treatment, for example, with immunosuppressive agents, which may risk the life of patients with HLS secondary to VL [174,175].
Amyloidosis has been rarely described in human patients with VL, except in a few cases of HIV co-infected patients. These patients develop renal failure due to amyloidosis with elevated levels of serum amyloid. Serum amyloid is one of the positive APRs. Amyloidosis also has been described in dogs and hamsters with VL [140,176,177].
6. Complications
Hemorrhage is a frequent clinical manifestation often with the severe consequence of death in VL. The severity varies from minor signs and symptoms, e.g., epistaxis, petechiae, and bruising to hemorrhage from the gums and urinary bleeding to open digestive hemorrhage and internal bleeding, followed by hypovolemic shock [58,87,89,90,99,100,101,102,103,107,108,109,110,111,112,127,146,159]. The underlying mechanisms of bleeding appears to be associated with DIC [104,159,160] plus a set of other anticoagulant changes. DIC is characterized by systemic activation of blood coagulation with production and deposition of fibrin that is followed by events leading to microvascular thrombi in various organs, hemorrhage or both, depending on the balance of coagulation cascade. Fibrinolysis and consumption of clotting factors (including platelets) can result in hemorrhage, which predominates over the thromboembolic phenomenon [178,179,180,181,182]. Overt and non-overt DIC are almost always present in VL, as noticed in the blood test by prolonged time of coagulation, high D-dimer, and low platelet counts [104]. DIC is triggered and fueled by the presence of proinflammatory cytokines, e.g., IL-6 and IL-1β, which increase the expression of tissue factor by the endothelium and mononuclear cells to elicit the extrinsic coagulation pathway [64,80,183].
Further evidence for a role of DIC is the presence of microthrombi in the liver of VL patients [125]. DIC is potentially driven indirectly by another mechanism via the overproduction of coagulation proteins by the liver secondary to the IL-6 driven APR [80]. A slight reduction in liver function may increase the chance of bleeding when DIC is already present [184], as noted with a modest rise in the blood level of liver enzymes, i.e., ALT and AST in VL. The retention of bilirubin in this disease may also reduce the availability of vitamin-K-dependent coagulation factors [185]. Finally, hypersplenism-dependent development of milder thrombocytopenia has the potential to predispose the patients to hemorrhage when other mechanisms of fibrinolysis have already set in.
Bacterial co-infection is the most frequent cause of death and thus one of the riskiest manifestations in VL [58,86,87,89,101,107,108,109,146]. The co-infection can be caused by either Gram-positive or Gram-negative bacteria [169,186,187]. The coexistence of tuberculosis with VL has been reported in some parts of the world, especially in the cases of HIV co-infection [47,88,90,101,102]. Acute bacterial nosocomial infection is another risk factor due to the prolonged in-hospital stay of VL patients [186]. Their propensity to secondary bacterial infection is far from having a clear-cut explanation. Some possibilities raised include lymphocyte anergy following exaggerated regulatory cytokine response, spleen disorganization, and malnutrition. The cause-and-effect relationship has not been well-established in the case of HIV co-infection as a risk factor for susceptibility of patients to severe secondary bacterial infection in VL. We have previously demonstrated that clinically, severe VL has strong similarities with bacterial sepsis in having a wave of proinflammatory cytokines leading to severe complications and mortality, albeit the systemic inflammatory response being slow-progressing or protracted [57]. We refer to this as leishmanial sepsis [188,189,190], lacking the acute components of bacterial sepsis, like time-dependent hypovolemic shock and the solid regulatory component. The latter renders patients of bacterial sepsis unable to contain the active infection and predisposes them to new bacterial, viral, or fungal infections [191,192,193,194]. Figure 3 depicts the progression of VL towards lethality.
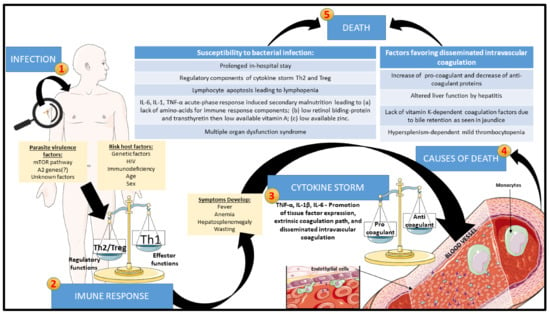
Figure 3.
Proposed evolution of VL to the death of the patients through leishmanial sepsis: After Leishmania infection, the progression of the disease can be exacerbated by interactions between parasite virulence and host factors. With the development of clinical signs and symptoms, leishmanial sepsis may ensue due to exaggerated innate immune response, which promotes increased tissue-factor expression by endothelium and monocytes, generating disseminated intravascular coagulation (DIC). At the end stage, DIC can trigger bleeding enhanced by acute phase reactions with elevation of procoagulant proteins and reduction in anticoagulant proteins, with liver function and bile retention being altered, reducing vitamin-K-dependent coagulation factors and thrombocytopenia secondary to hypersplenism. Simultaneously, prolonged hospital stays of the patients in conjunction with their lymphopenia and cachexia due to the production of IL-6, IL-1, and TNF-α and deprivation of amino acids, retinol, and zinc increase their susceptibility to bacterial infection and superimposed bacterial sepsis. These episodes of leishmanial sepsis fuel multiple organ dysfunction syndrome and death of the patients.
VL is indeed similar to bacterial sepsis in their proinflammatory and regulatory cytokine profiles. These cytokines are elevated in VL [51] and reach an extremely high level in severe disease [104,120]. One critical point of bacterial sepsis is the development of the so-called counter-inflammatory response syndrome (CARS), in which regulatory cells, cytokines (e.g., IL-10 and TGF-β), and other soluble factors inhibit the action of IFN-γ to mediate the mechanism of killing intracellular pathogens. This process is also called lymphocyte anergy due to the apoptosis of proinflammatory immune cells [191,193,194,195]. Similarly, elaboration of IL-10 and TGF-β is associated with an increased parasite burdens seen in patients with VL. Thus, CARS also occurs in VL, as a possible factor predisposing patients to the complication of bacterial infection and death.
Another potential factor predisposing patients to bacterial superinfection is lymphopenia. Indeed, some patients with bacterial sepsis suffer from profound lymphopenia, a marker of poor prognosis [196,197,198]. It has been proposed that apoptosis of lymphocytes plays a significant pathogenetic role in septic lymphopenia [199]. Moreover, patients with sepsis who received the human recombinant growth factor for the lymphoid lineage IL-7 CYT107 recovered from lymphopenia [200]. Lymphopenia was accounted for by a reduced thymic output of lymphocytes due to their apoptosis in VL-HIV co-infection [201,202]. Apoptosis of immune cells has been long described to play a role in human and animal infection and immunity [203,204]. To our knowledge, lymphopenia was proposed only in a single study as a risk factor for the death of patients with VL. Reanalysis of our previously published data [109] (Table 2) revealed that the mean and median lymphocyte counts were significantly lower in patients who died of VL than those who survived. Thus, lymphocyte depletion appears to increase the chance of secondary bacterial infections in VL as a significant cause of death. This finding raises the consideration of adjuvant therapy with recombinant IL-7 or check-point inhibitors for treating patients of VL with severe lymphopenia. While neutropenia did not contribute to the death of patients in our cases (Table 2), extreme neutropenia was noted to increase the risk of bacterial infections in other patients with the disease [58].
Table 2.
Disparity of dead versus survived patients of VL in their blood lymphocyte and neutrophil counts *.
Malnutrition is another factor which has been considered to predispose VL patients to bacterial infections. Undernutrition has been suggested to be a risk factor for developing VL (primary malnutrition), although it is also known as a consequence of VL (secondary malnutrition). Prospective cohorts of children exposed to L. infantum have been evaluated for the effect of nutrition with equivocal outcome of whether malnutrition predisposes patients to the infection or to the development of disease [9,10]. Malnutrition has been well described and generally accepted as a risk factor for many other infections, e.g., infectious diarrhea and pneumonia [147,193]. The work in this area is inherently difficult in the attribution of a multitude of immunological defects found in primary or secondary malnutrition [205]. Malnutrition secondary to VL, as discussed earlier, is similar to that of bacterial sepsis, resulting from systemic inflammation and APR.
Interestingly, the better nourished a patient is, the lower the parasite burdens seen in patients during the progression of VL. It is possible that secondary malnutrition might also be immunosuppressive, leaving Leishmania proliferation unchecked [39,59]. While how secondary malnutrition affects human immunity to the parasitism is not known, it probably differs mechanistically from the primary malnutrition. For instance, proinflammatory cytokines seen in VL are known to act on the hypothalamus to drive the loss of appetite [67,91,92]. The dramatic shift-down of liver protein synthesis also noted in VL [68] may lead to immunosuppression for the following accounts. The reduced synthesis of albumin with the increasing levels of APR theoretically favors the host defense by freeing amino acids for the synthesis of immunity-related proteins, e.g., CRP and complement components. However, a deficiency in albumin reduces its important function as a carrier for the transport of micronutrients, like zinc. Likewise, other negative APR, such as transthyretin and retinol-binding protein, may reduce the availability of vitamin A for its essential function in the immune system, rendering patients with VL more susceptible to bacterial infections [70,85]. Due to the relevant role of malnutrition in immunity, its potential impact on chemotherapy is worthy of investigation.
In summary, the risk for bacterial infection in VL may include the following factors: prolonged in-hospital stay, HIV co-infection, regulatory cytokine immunosuppression, lymphopenia, T cell exhaustion, severe neutropenia, and malnutrition, the last one being the most important.
7. Death
It is noteworthy that among all the risk factors identified to account for the severity of VL, few are causally linked to death. Patients do not die or rarely die directly from the typical symptoms and signs of VL, i.e., fever, anemia, edema, liver failure, renal failure, hyperglobulinemia, hypoalbuminemia, vomiting, diarrhea, neutropenia, and/or thrombocytopenia. Vomiting and diarrhea may lead to fatal dehydration, hypovolemia and hydroelectrolytic imbalance, but these are uncommon or rare complications of VL. As discussed above, severe disease may lead to some degree of hepatitis and jaundice that, coupled with DIC, can mimic acute liver failure. This is, however, not the real cause of death, since typical fulminant hepatitis has not been observed in autopsies of patients with VL [119,125,141]. Acute and severe bacterial co-infection is considered to aggravate leishmanial sepsis, accelerating progression to bleeding. Therefore, inflammatory end-stage leishmanial and bacterial sepsis seem to be the main causes of death in VL, resulting from septic shock, disseminated microthrombi, and multiple organ dysfunction syndrome.
The central determinant of leishmanial sepsis in human VL proposed is exacerbated inflammation, followed by counter-inflammation and immunosuppression. Many questions regarding this proposal await further investigation. Is it primarily dependent on the human host’s variabilities, such as age, sex, HIV co-infection, and genetic background? Or are there parasite factors that trigger systemic inflammation? A high probability of 83% for an association of VL mortality with parasite genomes was suggested from analysis of the sequence data from 109 L. infantum isolates in conjunction with the clinical data [30]. Currently, we are expanding the database to provide statistical power with the hope to precisely identify Leishmania virulence factors in play for designing better pharmaceuticals and vaccines.
8. Concluding Remarks—Future Investigation and Clinical Managements
Early infection in human VL (and leishmaniasis in general) is a blind spot in our knowledge from the moment of the infective transmission by sand fly bite to the time when patients seek medical attention. The very early symptoms, e.g., loss of appetite and apathy, are too vague for the patients to recall the time of their onset. Fever is thus usually taken as the first sign of this disease for the ease of its measurement. Ill-defined onset of VL generates bias in longitudinal studies for assessing risk factors, such as malnutrition. To minimize this and other uncertainties requires the information for the timeframe of transition from the very initial infection to the development of human disease. Such knowledge is expected to inform us about the equilibrium between susceptibility and resistance as well as how human immunity develops.
New approaches have been proposed for the clinical management of VL based on the available knowledge of its pathogenesis presented in Table 3. Supportive therapies and assessments of biomarkers are included with the aim to improve the outcome of severely ill patients. Since IL-6 triggers DIC and the acute phase response central to the pathogenesis of VL, anti-IL-6 and its receptor antagonists are worthy of consideration to use for treating patients with cytokine storm. Evaluation of efficacy is needed after short-term and longer-term treatments. Recombinant antithrombin concentrates and recombinant thrombomodulin have been used to treat bacterial sepsis for DIC of the suppressed fibrinolytic type. These drugs may be used for VL with care due to bleeding complications. Also worthy of consideration for proof of concept is to treat VL with tranexamic acid, an antifibrinolytic drug, which has been widely used for several different clinical conditions, including bacterial sepsis. Vitamin K must be evaluated for treating VL patients with jaundice and hemorrhage. Tests for DIC polarization are recommended for investigation to optimize anti-DIC therapies in VL. It is feasible to assess IL-10 and TGF-beta as potential risk factors and as targets for neutralization to mitigate bacterial sepsis associated with VL. VL patients with absolute lymphopenia offer an excellent opportunity for testing recombinant IL-7 and immune checkpoint inhibitors to reverse lymphocyte depletion. Finally, the early use of antibiotics needs to be assessed by clinical trials, particularly when specific biomarkers are detected, indicative of bacterial sepsis [206].
Table 3.
Proposed new approaches to clinical management of visceral leishmaniasis.
How noninfected organs, like lungs, liver, and kidney are involved in the pathology of VL is largely unknown. The inflammatory trigger of DIC as a cause remains speculative. Further investigation of the infected organs, e.g., bone marrow, spleen, and liver is also desirable for understanding their susceptibility to the infection and their relative roles in host defense. The slow progression of cytokine storm in VL is of special interest for detailed investigation, as the outcome may shed light on similar events in other infectious diseases with systemic inflammation profiles, like bacterial sepsis, COVID-19, and particularly bacterial superinfection and multiple organ dysfunction syndrome. Of particular relevance for such investigation is the use of CHIM, which is apparently underway to assess vaccines of different formats against L. major and L. donovani infection challenges [26,207]. Other emerging tools of value include 3D organoid culture involving the use of human cells with different lineages [208] like immune cells, endothelium, hepatocytes, and neurons. All these new approaches are expected to become available to provide means to deal with severe disease more effectively, raising our hope for a reversal of the trend toward increasing mortality as seen in urban VL in Brazil.
Author Contributions
C.H.N.C., conceptualization, writing, and editing; K.-P.C., review and writing; D.L.C., patients’ images and editing; F.V.C.C., graphic abstract, Figure 3, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
CHN Costa has a Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) fellowship, grant number 307606/221-0.
Institutional Review Board Statement
There are no ethical conflicts to declare.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kassebaum, N.J.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, J.; Carter, A.; Casey, D.C.; Charlson, F.J.; Coates, M.M.; Coggeshall, M.; et al. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1603–1658. [Google Scholar] [CrossRef]
- Wamai, R.G.; Kahn, J.; McGloin, J.; Ziaggi, G. Visceral leishmaniasis: A global overview. J. Glob. Health Sci. 2020, 2, e3. [Google Scholar] [CrossRef]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mandal, R.; Das, S.; Kesari, S.; Dinesh, D.S.; Pandey, K.; Das, V.R.; Topno, R.K.; Sharma, M.P.; Dasgupta, R.K.; et al. Kala-azar elimination in a highly-endemic district of Bihar, India: A success story. PLoS Negl. Trop. Dis. 2020, 14, e0008254. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Kala-azar in India—progress and challenges towards its elimination as a public health problem. Wkly Epidemiol Rec. 2021, 96, 267–279. [Google Scholar]
- Cota, G.; Erber, A.C.; Schernhammer, E.; Simões, T.C. Inequalities of visceral leishmaniasis case-fatality in Brazil: A multilevel modeling considering space, time, individual and contextual factors. PLoS Negl. Trop. Dis. 2021, 15, e0009567. [Google Scholar] [CrossRef]
- Pampiglione, S.; Manson-Bahr, P.E.; La Placa, M.; Borgatti, M.A.; Musumeci, S. Studies in Mediterranean leishmaniasis. 3. The leishmanin skin test in kala-azar. Trans. R. Soc. Trop. Med. Hyg. 1975, 69, 60–68. [Google Scholar] [CrossRef]
- Badaro, R.; Jones, T.C.; Lorenco, R.; Cerf, B.J.; Sampaio, D.; Carvalho, E.M.; Rocha, H.; Teixeira, R.; Johnson, W.D. A Prospective Study of Visceral Leishmaniasis in an Endemic Area of Brazil. J. Infect. Dis. 1986, 154, 639–649. [Google Scholar] [CrossRef]
- Evans, T.G.; Teixeira, M.J.; McAuliffe, I.T.; Vasconcelos, I.; Vasconcelos, A.W.; Sousa, A.d.A.; Lima, J.W.; Pearson, R.D. Epidemiology of visceral leishmaniasis in northeast Brazil. J. Infect. Dis. 1992, 166, 1124–1132. [Google Scholar] [CrossRef]
- Carstens-Kass, J.; Paulini, K.; Lypaczewski, P.; Matlashewski, G. A review of the leishmanin skin test: A neglected test for a neglected disease. PLoS Negl. Trop. Dis. 2021, 15, e0009531. [Google Scholar] [CrossRef] [PubMed]
- Werneck, G.L.; Rodrigues, L.; Santos, M.V.; Araújo, I.B.; Moura, L.S.; Lima, S.S.; Gomes, R.B.B.B.; Maguire, J.H.; Costa, C.H.H.N. The burden of Leishmania chagasi infection during an urban outbreak of visceral leishmaniasis in Brazil. Acta Trop. 2002, 83, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, M.; Moreno, I.; Aizpurua, C.; Toraño, A. Early mechanisms of Leishmania infection in human blood. Microbes Infect. 2003, 5, 507–513. [Google Scholar] [CrossRef]
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 2008, 321, 970–974. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Nemati, M.; Sharifi, I.; Nair, A.; Shukla, D.; Chauhan, P.; Khorramdelazad, H.; Sarkar, A.; Saha, B. Leishmania species-dependent functional duality of toll-like receptor 2. IUBMB Life 2019, 71, 1685–1700. [Google Scholar] [CrossRef]
- Wu, T.-H.; Hsieh, S.-C.; Li, T.-H.; Lu, C.-H.; Liao, H.-T.; Shen, C.-Y.; Li, K.-J.; Wu, C.-H.; Kuo, Y.-M.; Tsai, C.-Y.; et al. Molecular Basis for Paradoxical Activities of Polymorphonuclear Neutrophils in Inflammation/Anti-Inflammation, Bactericide/Autoimmunity, Pro-Cancer/Anticancer, and Antiviral Infection/SARS-CoV-II-Induced Immunothrombotic Dysregulation. Biomedicines 2022, 10, 773. [Google Scholar] [CrossRef]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Loré, K. Neutrophils acquire the capacity for antigen presentation to memory CD4+ T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef]
- van Zandbergen, G.; Solbach, W.; Laskay, T. Apoptosis driven infection. Autoimmunity 2007, 40, 349–352. [Google Scholar] [CrossRef]
- Volpedo, G.; Pacheco-Fernandez, T.; Holcomb, E.A.; Cipriano, N.; Cox, B.; Satoskar, A.R. Mechanisms of Immunopathogenesis in Cutaneous Leishmaniasis And Post Kala-azar Dermal Leishmaniasis (PKDL). Front. Cell. Infect. Microbiol. 2021, 11, 685296. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Fasel, N. How to master the host immune system? Leishmania parasites have the solutions! Int. Immunol. 2018, 30, 103–111. [Google Scholar] [CrossRef]
- Tibúrcio, R.; Nunes, S.; Nunes, I.; Rosa Ampuero, M.; Silva, I.B.; Lima, R.; Machado Tavares, N.; Brodskyn, C. Molecular Aspects of Dendritic Cell Activation in Leishmaniasis: An Immunobiological View. Front. Immunol. 2019, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.J.C.; Zacarias, D.A.D.A.; Silva, V.C.V.C.; Rolão, N.; Costa, D.L.D.L.; Costa, C.H.C.H.N. Comparison of optical microscopy and quantitative polymerase chain reaction for estimating parasitaemia in patients with kala-azar and modelling infectiousness to the vector Lutzomyia longipalpis. Mem. Inst. Oswaldo Cruz 2016, 111, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Kumar, R.; Singh, N.; Singh, A.K.; Rai, M.; Sacks, D.; Sundar, S.; Nylén, S. CD8 T cell exhaustion in human visceral leishmaniasis. J. Infect. Dis. 2014, 209, 290–299. [Google Scholar] [CrossRef]
- Bhor, R.; Rafati, S.; Pai, K. Cytokine saga in visceral leishmaniasis. Cytokine 2020, 147, 155322. [Google Scholar] [CrossRef]
- Volpedo, G.; Bhattacharya, P.; Gannavaram, S.; Pacheco-Fernandez, T.; Oljuskin, T.; Dey, R.; Satoskar, A.R.; Nakhasi, H.L. The History of Live Attenuated Centrin Gene-Deleted Leishmania Vaccine Candidates. Pathogens 2022, 11, 431. [Google Scholar] [CrossRef]
- Kolářová, I.; Valigurová, A. Hide-and-Seek: A Game Played between Parasitic Protists and Their Hosts. Microorganisms 2021, 9, 2434. [Google Scholar] [CrossRef]
- Lypaczewski, P.; Zhang, W.-W.; Matlashewski, G. Evidence that a naturally occurring single nucleotide polymorphism in the RagC gene of Leishmania donovani contributes to reduced virulence. PLoS Negl. Trop. Dis. 2021, 15, e0009079. [Google Scholar] [CrossRef]
- Kariyawasam, K.K.G.D.U.L.; Selvapandiyan, A.; Siriwardana, H.V.Y.D.; Dube, A.; Karunanayake, P.; Senanayake, S.A.S.C.; DEY, R.; Gannavaram, S.; NAKHASI, H.L.; KARUNAWEERA, N.D. Dermotropic Leishmania donovani in Sri Lanka: Visceralizing potential in clinical and preclinical studies. Parasitology 2018, 145, 443–452. [Google Scholar] [CrossRef]
- Grace, C.A.; Sousa Carvalho, K.S.; Sousa Lima, M.I.; Costa Silva, V.; Reis-Cunha, J.L.; Brune, M.J.; Forrester, S.; Pedrozo e Silva de Azevedo, C.d.M.; Costa, D.L.; Speed, D.; et al. Parasite Genotype Is a Major Predictor of Mortality from Visceral Leishmaniasis. MBio 2022, 13, e0206822. [Google Scholar] [CrossRef]
- Killingley, B.; Mann, A.J.; Kalinova, M.; Boyers, A.; Goonawardane, N.; Zhou, J.; Lindsell, K.; Hare, S.S.; Brown, J.; Frise, R.; et al. Safety, tolerability and viral kinetics during SARS-CoV-2 human challenge in young adults. Nat. Med. 2022, 28, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Martinez, E.; Suazo-Sanchez, I.; Celis-Romero, M.; Carnero, A. 3D and organoid culture in research: Physiology, hereditary genetic diseases and cancer. Cell Biosci. 2022, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Chan Zuckerberg Initiative. Available online: https://chanzuckerberg.com/newsroom/biohub-chicago-engineer-technologies-measure-human-biology (accessed on 24 April 2023).
- Bucheton, B.; Argiro, L.; Chevillard, C.; Marquet, S.; Kheir, M.M.; Mergani, A.; El-Safi, S.H.; Dessein, A.J. Identification of a novel G245R polymorphism in the IL-2 receptor β membrane proximal domain associated with human visceral leishmaniasis. Genes Immun. 2007, 8, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.P.; Ferreira, A.F.; Ribolla, P.E.; de Miranda Santos, I.K.; do Socorro Pires e Cruz, M.; Aécio de Carvalho, F.; Abatepaulo, A.R.; Lamounier Costa, D.; Werneck, G.L.; Farias, T.J.; et al. Genotypes of the Mannan-Binding Lectin Gene and Susceptibility to Visceral Leishmaniasis and Clinical Complications. J. Infect. Dis. 2007, 195, 1212–1217. [Google Scholar] [CrossRef]
- Frade, A.F.; Oliveira, L.C.; Costa, D.L.; Costa, C.H.; Aquino, D.; Van Weyenbergh, J.; Barral-Netto, M.; Barral, A.; Kalil, J.; Goldberg, A.C. TGFB1 and IL8 gene polymorphisms and susceptibility to visceral leishmaniasis. Infect. Genet. Evol. 2011, 11, 912–916. [Google Scholar] [CrossRef]
- Blackwell, J.M.; Fakiola, M.; Castellucci, L.C. Human genetics of leishmania infections. Hum. Genet. 2020, 139, 813–819. [Google Scholar] [CrossRef]
- Caldas, A.J.M.; Costa, J.M.L.; Silva, A.A.M.; Vinhas, V.; Barral, A. Risk factors associated with asymptomatic infection by Leishmania chagasi in north-east Brazil. Trans. R. Soc. Trop. Med. Hyg. 2002, 96, 21–28. [Google Scholar] [CrossRef]
- Zacarias, D.A.; Rolão, N.; de Pinho, F.A.; Sene, I.; Silva, J.C.; Pereira, T.C.; Costa, D.L.; Costa, C.H.N. Causes and consequences of higher Leishmania infantum burden in patients with kala-azar: A study of 625 patients. Trop. Med. Int. Health 2017, 22, 679–687. [Google Scholar] [CrossRef]
- Yan-jia, L. A review of kala-azar in China from 1949 to 1959. Trans. R. Soc. Trop. Med. Hyg. 1982, 76, 531–537. [Google Scholar] [CrossRef]
- Costa, C.H.N.; Pereira, H.F.; Araújo, M.V. Epidemia de leishmaniose visceral no Estado do Piauí, Brasil, 1980–1986. Rev. Saude Publica 1990, 24, 361–372. [Google Scholar] [CrossRef]
- Sharma, M.C.; Gupta, A.K.; Das, V.N.; Verma, N.; Kumar, N.; Saran, R.; Kar, S.K. Leishmania donovani in blood smears of asymptomatic persons. Acta Trop. 2000, 76, 195–196. [Google Scholar] [CrossRef]
- Cloots, K.; Burza, S.; Malaviya, P.; Hasker, E.; Kansal, S.; Mollett, G.; Chakravarty, J.; Roy, N.; Lal, B.K.; Rijal, S.; et al. Male predominance in reported Visceral Leishmaniasis cases: Nature or nurture? A comparison of population-based with health facility-reported data. PLoS Negl. Trop. Dis. 2020, 14, e0007995. [Google Scholar] [CrossRef]
- Araújo Albuquerque, L.P.; Silva, A.M.; Araújo Batista, F.M.; Souza Sene, I.; Costa, D.L.; Costa, C.H.N.; de Araújo Albuquerque, L.P.; da Silva, A.M.; de Araújo Batista, F.M.; de Souza Sene, I.; et al. Influence of sex hormones on the immune response to leishmaniasis. Parasite Immunol. 2021, 43, e12874. [Google Scholar] [CrossRef] [PubMed]
- Khalil, E.A.G.; Zijlstra, E.E.; Kager, P.A.; El Hassan, A.M. Epidemiology and clinical manifestations of Leishmania donovani infection in two villages in an endemic area in eastern Sudan. Trop. Med. Int. Health 2002, 7, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.S.; Barreto, A.S.; Bomfim, L.G.S.; Gomes, M.C.; Ferreira, N.L.C.; da Cruz, G.S.; Magalhães, L.S.; de Jesus, A.R.; Palatnik-de-Sousa, C.B.; Corrêa, C.B.; et al. Multifunctional, TNF-α and IFN-γ-Secreting CD4 and CD8 T Cells and CD8High T Cells Are Associated With the Cure of Human Visceral Leishmaniasis. Front. Immunol. 2021, 12, 773983. [Google Scholar] [CrossRef] [PubMed]
- Akuffo, H.; Costa, C.; van Griensven, J.; Burza, S.; Moreno, J.; Herrero, M. New insights into leishmaniasis in the immunosuppressed. PLoS Negl. Trop. Dis. 2018, 12, e0006375. [Google Scholar] [CrossRef]
- Serafim, T.D.; Coutinho-Abreu, I.V.; Dey, R.; Kissinger, R.; Valenzuela, J.G.; Oliveira, F.; Kamhawi, S. Leishmaniasis: The act of transmission. Trends Parasitol. 2021, 37, 976–987. [Google Scholar] [CrossRef]
- Kimblin, N.; Peters, N.; Debrabant, A.; Secundino, N.; Egen, J.; Lawyer, P.; Fay, M.P.; Kamhawi, S.; Sacks, D. Quantification of the infectious dose of Leishmania major transmitted to the skin by single sand flies. Proc. Natl. Acad. Sci. USA 2008, 105, 10125–10130. [Google Scholar] [CrossRef]
- Giraud, E.; Martin, O.; Yakob, L.; Rogers, M. Quantifying Leishmania Metacyclic Promastigotes from Individual Sandfly Bites Reveals the Efficiency of Vector Transmission. Commun. Biol. 2019, 2, 84. [Google Scholar] [CrossRef]
- Peruhype-Magalhaes, V.; Martins-Filho, O.A.; Prata, A.; Silva, L.D.A.; Rabello, A.; Teixeira-Carvalho, A.; Figueiredo, R.M.; Guimarães-Carvalho, S.F.; Ferrari, T.C.A.; Van Weyenbergh, J.; et al. Mixed inflammatory/regulatory cytokine profile marked by simultaneous raise of interferon-gamma and interleukin-10 and low frequency of tumour necrosis factor-alpha(+) monocytes are hallmarks of active human visceral Leishmaniasis due to Leishmania chagas. Clin. Exp. Immunol. 2006, 146, 124–132. [Google Scholar] [CrossRef]
- Jawed, J.J.; Dutta, S.; Majumdar, S. Functional aspects of T cell diversity in visceral leishmaniasis. Biomed. Pharmacother. 2019, 117, 109098. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Bacellar, O.; Barral, A.; Badaro, R.; Johnson, W.D.; Carvalho, E.M.; Bacellar, O.; Barral, A.; Badaro, R.; Johnson, W.D.; et al. Antigen-specific immunosuppression in visceral leishmaniasis is cell mediated. J. Clin. Investig. 1989, 83, 860–864. [Google Scholar] [CrossRef]
- Caldas, A.; Favali, C.; Aquino, D.; Vinhas, V.; van Weyenbergh, J.; Brodskyn, C.; Costa, J.; Barral-Netto, M.; Barral, A. Balance of IL-10 and interferon-gamma plasma levels in human visceral leishmaniasis: Implications in the pathogenesis. BMC Infect. Dis. 2005, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Nylén, S.; Maurya, R.; Eidsmo, L.; Das Manandhar, K.; Sundar, S.; Sacks, D. Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J. Exp. Med. 2007, 204, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, E.M.; Badaró, R.; Reed, S.G.; Jones, T.C.; Johnson, W.D. Absence of gamma interferon and interleukin 2 production during active visceral leishmaniasis. J. Clin. Investig. 1985, 76, 2066–2069. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Teixeira, R.S.; Johnson, W.D. Cell-mediated immunity in American visceral leishmaniasis: Reversible immunosuppression during acute infection. Infect. Immun. 1981, 33, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.H.N.; Werneck, G.L.; Costa, D.L.; Holanda, T.A.; Aguiar, G.B.; Carvalho, A.S.; Cavalcanti, J.C.; Santos, L.S. Is severe visceral leishmaniasis a systemic inflammatory response syndrome? A case control study. Rev. Soc. Bras. Med. Trop. 2010, 43, 386–392. [Google Scholar] [CrossRef]
- Costa, C.H.N.; Zacarias, D.A.; Soares, M.R.A.; Silva, J.M.; Costa, D.L.; de Figueirêdo, L.C.; Ishikawa, E.A.Y. Bone Marrow Parasite Burden among Patients with New World Kala-Azar is Associated with Disease Severity. Am. J. Trop. Med. Hyg. 2014, 90, 621–626. [Google Scholar] [CrossRef]
- Chauhan, P.; Shukla, D.; Chattopadhyay, D.; Saha, B. Redundant and regulatory roles for Toll-like receptors in Leishmania infection. Clin. Exp. Immunol. 2017, 190, 167–186. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.-J.J.; Ott, I.; Marx, N.; Luther, T.; Kenngott, S.; Gawaz, M.; Kotzsch, M.; Schömig, A.; Schömig, A. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3399–3405. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, L.; Zhao, Y.; Hara, H.; Chen, P.; Xu, J.; Tang, J.; Wei, L.; Li, Z.; Cooper, D.K.C.; et al. Human IL-6, IL-17, IL-1β, and TNF-α differently regulate the expression of pro-inflammatory related genes, tissue factor, and swine leukocyte antigen class I in porcine aortic endothelial cells. Xenotransplantation 2017, 24, e12291. [Google Scholar] [CrossRef] [PubMed]
- Vallières, L.; Rivest, S. Regulation of the Genes Encoding Interleukin-6, Its Receptor, and gp130 in the Rat Brain in Response to the Immune Activator Lipopolysaccharide and the Proinflammatory Cytokine Interleukin-1β. J. Neurochem. 1997, 69, 1668–1683. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2016, 54, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Welte, T.; Mahadeva, R. Acute Phase Proteins: Structure and Function Relationship [Internet]. In Acute Phase Proteins—Regulation and Functions of Acute Phase Proteins; Veas, F., Ed.; In Tech: London, UK, 2011; p. 368. ISBN 978-953-307-252-4. [Google Scholar]
- Markanday, A. Acute Phase Reactants in Infections: Evidence-Based Review and a Guide for Clinicians. Open Forum Infect. Dis. 2015, 2, ofv098. [Google Scholar] [CrossRef]
- Rasic, I.; Rebic, V.; Rasic, A.; Aksamija, G.; Radovic, S. The Association of Simultaneous Increase in Interleukin-6, C Reactive Protein, and Matrix Metalloproteinase-9 Serum Levels with Increasing Stages of Colorectal Cancer. J. Oncol. 2018, 2018, 2830503. [Google Scholar] [CrossRef]
- Wasunna, K.M.; Raynes, J.G.; Were, J.B.; Muigai, R.; Sherwood, J.; Gachihi, G.; Carpenter, L.; McAdam, K.P. Acute phase protein concentrations predict parasite clearance rate during therapy for visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 1995, 89, 678–681. [Google Scholar] [CrossRef]
- Singh, U.K.; Patwari, A.K.; Sinha, R.K.; Kumar, R. Prognostic value of serum C-reactive protein in kala-azar. J. Trop. Pediatr. 1999, 45, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.A.; Sharma, P.; Salotra, P. Circulating nitric oxide and C-reactive protein levels in Indian kala azar patients: Correlation with clinical outcome. Clin. Immunol. 2007, 122, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.N.N.P.; de Almeida, A.T.A.; Costa, D.L.; Costa, C.H.N. C-reactive protein for the diagnosis and prognosis of visceral leishmaniasis caused by Leishmania infantum. J. Trop. Pathol. 2022, 51, 145–156. [Google Scholar]
- Endale, H.T.; Mengstie, T.A.; Dawit, D.D.; Mohammed, R.; Dessie, G.; Tesfa, K.H. Assessment of liver function test and associated factors among visceral leishmaniasis patients attending university of gondar leishmaniasis research and treatment center, Northwest Ethiopia. PLoS ONE 2021, 16, e0260022. [Google Scholar] [CrossRef]
- Varma, N.; Naseem, S. Hematologic changes in visceral leishmaniasis/kala azar. Indian J. Hematol. Blood Transfus. 2010, 26, 78–82. [Google Scholar] [CrossRef]
- Bray, C.; Bell, L.N.; Liang, H.; Haykal, R.; Kaiksow, F.; Mazza, J.J.; Yale, S.H. Erythrocyte Sedimentation Rate and C-reactive Protein Measurements and Their Relevance in Clinical Medicine. WMJ 2016, 115, 317–321. [Google Scholar]
- Tishkowski, K.; Gupta, V. Erythrocyte Sedimentation Rate; In Tech: London, UK, 2022. [Google Scholar]
- Davidson, S.J. Inflammation and Acute Phase Proteins in Haemostasis. In Acute Phase Proteins; In Tech: London, UK, 2013; pp. 31–54. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.-F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Roth, J.; Blatteis, C.M. Mechanisms of fever production and lysis: Lessons from experimental LPS fever. Compr. Physiol. 2014, 4, 1563–1604. [Google Scholar] [CrossRef]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Rijal, S.; Chappuis, F.; Sundar, S.; Lima, M.A.; Olliaro, P.L.; Costa, C.H.; Vaillant, M.; Costa, D.L.; Balasegaram, M.; Harhay, M.O.; et al. Who Is a Typical Patient with Visceral Leishmaniasis? Characterizing the Demographic and Nutritional Profile of Patients in Brazil, East Africa, and South Asia. Am. J. Trop. Med. Hyg. 2011, 84, 543–550. [Google Scholar] [CrossRef]
- Monteiro, M.J.S.D.; Silva, M.N.P.D.; Paiva, A.A.; Marreiro, D.D.N.; Luzia, L.A.; Henriques, G.S.; Rondó, P.H.C.; Sene, I.S.; Almeida, A.T.A.; Costa, C.H.N.; et al. Nutritional status and vitamin A and zinc levels in patients with kala-azar in Piauí, Brazil. Rev. Soc. Bras. Med. Trop. 2021, 54, e08002020. [Google Scholar] [CrossRef] [PubMed]
- Seaman, J.; Mercer, A.J.; Sondorp, H.E.; Herwaldt, B.L. Epidemic Visceral Leishmaniasis in Southern Sudan: Treatment of Severely Debilitated Patients under Wartime Conditions and with Limited Resources. Ann. Intern. Med. 1996, 124, 664. [Google Scholar] [CrossRef] [PubMed]
- Rey, L.C.; Martins, C.V.; Ribeiro, H.B.; Lima, A.A.M. American visceral leishmaniasis (kala-azar) in hospitalized children from an endemic area. J. Pediatr. 2005, 81, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Herrero, M.; Orfanos, G.; Argaw, D.; Mulugeta, A.; Aparicio, P.; Parreño, F.; Bernal, O.; Rubens, D.; Pedraza, J.; Lima, M.A.; et al. Natural history of a visceral leishmaniasis outbreak in highland Ethiopia. Am. J. Trop. Med. Hyg. 2009, 81, 373–377. [Google Scholar] [CrossRef] [PubMed]
- da Silva, T.A.M.; Morais, M.H.F.; Lopes, H.M.d.O.R.; Gonçalves, S.A.; Magalhães, F.d.C.; Amâncio, F.F.; Antunes, C.M.F.; Carneiro, M.; de Oliveira Ramos Lopes, H.M.; Gonçalves, S.A.; et al. Prognostic factors associated with death from visceral leishmaniasis: A case-control study in Brazil. Trans. R. Soc. Trop. Med. Hyg. 2020, 114, 346–354. [Google Scholar] [CrossRef]
- Abongomera, C.; van Henten, S.; Vogt, F.; Buyze, J.; Verdonck, K.; van Griensven, J. Prognostic factors for mortality among patients with visceral leishmaniasis in East Africa: Systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2020, 14, e0008319. [Google Scholar] [CrossRef]
- Thaler, J.P.; Choi, S.J.; Schwartz, M.W.; Wisse, B.E. Hypothalamic inflammation and energy homeostasis: Resolving the paradox. Front. Neuroendocrinol. 2010, 31, 79–84. [Google Scholar] [CrossRef]
- Harden, L.M.; Kent, S.; Pittman, Q.J.; Roth, J. Fever and sickness behavior: Friend or foe? Brain. Behav. Immun. 2015, 50, 322–333. [Google Scholar] [CrossRef]
- Rothhaas, R.; Chung, S. Role of the Preoptic Area in Sleep and Thermoregulation. Front. Neurosci. 2021, 15, 664781. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.; Fearon, K.C. Cachexia, survival and the acute phase response. Curr. Opin. Support. Palliat. Care 2008, 2, 267–274. [Google Scholar] [CrossRef]
- Carson, J.A.; Baltgalvis, K.A. Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc. Sport Sci. Rev. 2010, 38, 168–176. [Google Scholar] [CrossRef]
- Narsale, A.A.; Carson, J.A. Role of interleukin-6 in cachexia: Therapeutic implications. Curr. Opin. Support. Palliat. Care 2014, 8, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Scorza, K.; Williams, A.; Phillips, J.D.; Shaw, J. Evaluation of nausea and vomiting. Am. Fam. Physician 2007, 76, 76–84. [Google Scholar] [PubMed]
- Singh, P.; Yoon, S.S.; Kuo, B. Nausea: A review of pathophysiology and therapeutics. Therap. Adv. Gastroenterol. 2016, 9, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.; Veeken, H.; Long, J. Visceral leishmaniasis and HIV in Tigray, Ethiopia. Trop. Med. Int. Health 2003, 8, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Collin, S.; Davidson, R.; Ritmeijer, K.; Keus, K.; Melaku, Y.; Kipngetich, S.; Davies, C. Conflict and Kala-Azar: Determinants of Adverse Outcomes of Kala-Azar among Patients in Southern Sudan. Clin. Infect. Dis. 2004, 38, 612–619. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, V.E.M.; Morais, M.H.F.; Reis, I.A.; Rabello, A.; Carneiro, M. Early Clinical Manifestations Associated with Death from Visceral Leishmaniasis. PLoS Negl. Trop. Dis. 2012, 6, e1511. [Google Scholar] [CrossRef]
- Madalosso, G.; Fortaleza, C.M.; Ribeiro, A.F.; Cruz, L.L.; Nogueira, P.A.; Lindoso, J.A. American Visceral Leishmaniasis: Factors Associated with Lethality in the State of São Paulo, Brazil. J. Trop. Med. 2012, 2012, 281572. [Google Scholar] [CrossRef]
- Belo, V.S.; Struchiner, C.J.; Barbosa, D.S.; Nascimento, B.W.L.; Horta, M.A.P.; da Silva, E.S.; Werneck, G.L. Risk Factors for Adverse Prognosis and Death in American Visceral Leishmaniasis: A Meta-analysis. PLoS Negl. Trop. Dis. 2014, 8, e2982. [Google Scholar] [CrossRef]
- Costa, D.L.; Rocha, R.L.; Carvalho, R.M.A.; Lima-Neto, A.S.; Harhay, M.O.; Costa, C.H.N.; Barral-Neto, M.; Barral, A.P. Serum cytokines associated with severity and complications of kala-azar. Pathog. Glob. Health 2013, 107, 78–87. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Mathew, A.; Rothman, A.L. Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol. 2017, 39, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoula, M.S.; M’Hamdi, Z.; Amri, F.; Tebib, N.; Ben Turkia, H.; Ben Dridi, M.F. Visceral leishmaniasis in children: Prognostic factors. Tunis. Med. 2003, 81, 535–539. [Google Scholar] [PubMed]
- Werneck, G.L.L.; Batista, M.S.A.S.A.; Gomes, J.R.B.R.B.; Costa, D.L.L.; Costa, C.H.N.H.N. Prognostic Factors for Death from Visceral Leishmaniasis in Teresina, Brazil. Infection 2003, 31, 174–177. [Google Scholar] [CrossRef]
- Sampaio, M.J.A.D.Q.; Cavalcanti, N.V.; Alves, J.G.B.; Fernandes-Filho, M.J.C.; Correia, J.B. Risk factors for death in children with visceral leishmaniasis. PLoS Negl. Trop. Dis. 2010, 4, e877. [Google Scholar] [CrossRef] [PubMed]
- Coura-Vital, W.; de Araújo, V.E.M.; Reis, I.A.; Amancio, F.F.; Reis, A.B.; Carneiro, M. Prognostic Factors and Scoring System for Death from Visceral Leishmaniasis: An Historical Cohort Study in Brazil. PLoS Negl. Trop. Dis. 2014, 8, e3374. [Google Scholar] [CrossRef]
- Costa, D.L.; Rocha, R.L.; Chaves, E.d.B.F.; Batista, V.G.d.V.; Costa, H.L.; Costa, C.H.N. Predicting death from kala-azar: Construction, development, and validation of a score set and accompanying software. Rev. Soc. Bras. Med. Trop. 2016, 49, 728–740. [Google Scholar] [CrossRef]
- Ben Helel, K.; Ben Rejeb, M.; Habboul, Z.; Khattat, N.; Mejaouel, H.; Said-Latiri, H.; Kaabi, B.; Zhioua, E.; Helel, K.B.; Rejeb, M.B.; et al. Risk factors for mortality of children with zoonotic visceral leishmaniasis in Central Tunisia. PLoS ONE 2017, 12, e0189725. [Google Scholar] [CrossRef]
- Foinquinos, J.; Duarte, M.d.C.; Figueiroa, J.N.; Correia, J.B.; Cavalcanti, N.V. Temporal Validation of a Predictive Score for Death in Children with Visceral Leishmaniasis. J. Trop. Med. 2021, 2021, 6688444. [Google Scholar] [CrossRef]
- Kager, P.A.; Rees, P.H. Haematological investigations in visceral leishmaniasis. Trop. Geogr. Med. 1986, 38, 371–379. [Google Scholar]
- Boyer, T.D.; Habib, S. Big spleens and hypersplenism: Fix it or forget it? Liver Int. 2015, 35, 1492–1498. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Singh, S.S.; Sundar, S. Hepcidin mediated iron homoeostasis as immune regulator in visceral leishmaniasis patients. Parasite Immunol. 2019, 41, e12601. [Google Scholar] [CrossRef] [PubMed]
- Veress, B.; Omer, A.; Satir, A.A.; El Hassan, A.M. Morphology of the spleen and lymph nodes in fatal visceral leishmaniasis. Immunology 1977, 33, 605–610. [Google Scholar] [PubMed]
- Duarte, M.I.S.; Corbett, C.E.P. Histopathological patterns of the liver involvement in visceral leishmaniasis. Rev. Inst. Med. Trop. Sao Paulo 1987, 29, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Dos-Santos, W.L.; Pagliari, C.; Santos, L.G.; Almeida, V.A.; e Silva, T.L.; Coutinho, J.D., Jr.; Souza, T.; Duarte, M.I.; de Freitas, L.A.; Costa, C.H. A case of conventional treatment failure in visceral leishmaniasis: Leukocyte distribution and cytokine expression in splenic compartments. BMC Infect. Dis. 2014, 14, 491. [Google Scholar] [CrossRef]
- Hermida, M.D.; de Melo, C.V.B.; Lima, I.d.S.; Oliveira, G.G.d.S.; Dos-Santos, W.L.C. Histological Disorganization of Spleen Compartments and Severe Visceral Leishmaniasis. Front. Cell. Infect. Microbiol. 2018, 8, 394. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Ghosh, S.; Ejazi, S.A.; Rahaman, M.; Pandey, K.; Ravi Das, V.N.; Das, P.; Goswami, R.P.; Saha, B.; Ali, N. Induction of IL-10 and TGFβ from CD4+CD25+FoxP3+ T Cells Correlates with Parasite Load in Indian Kala-azar Patients Infected with Leishmania donovani. PLoS Negl. Trop. Dis. 2016, 10, e0004422. [Google Scholar] [CrossRef]
- Cavalcanti, A.S.; Ribeiro-Alves, M.; Pereira, L.D.O.R.; Mestre, G.L.; Ferreira, A.B.R.; Morgado, F.N.; Boité, M.C.; Cupolillo, E.; Moraes, M.O.; Porrozzi, R. Parasite Load Induces Progressive Spleen Architecture Breakage and Impairs Cytokine mRNA Expression in Leishmania infantum-Naturally Infected Dogs. PLoS ONE 2015, 10, e0123009. [Google Scholar] [CrossRef]
- Reinaldo, L.G.C.; Araújo-Júnior, R.J.C.; Diniz, T.M.; Moura, R.D.; Meneses-Filho, A.J.; Furtado, C.V.V.M.; Santos, W.L.C.; Costa, D.L.; Eulálio, K.D.; Ferreira, G.R.; et al. Splenectomy in Patients with Visceral Leishmaniasis Resistant to Conventional Therapy and Secondary Prophylaxis: A Retrospective Cohort. Am. J. Trop. Med. Hyg. 2022, in press. [Google Scholar] [CrossRef]
- el Hag, I.A.; Hashim, F.A.; el Toum, I.A.; Homeida, M.; el Kalifa, M.; el Hassan, A.M. Liver morphology and function in visceral leishmaniasis (Kala-azar). J. Clin. Pathol. 1994, 47, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Luz, K.G.; Tuon, F.F.; Duarte, M.I.S.; Maia, G.M.; Matos, P.; Ramos, A.M.d.O.; Nicodemo, A.C. Cytokine expression in the duodenal mucosa of patients with visceral leishmaniasis. Rev. Soc. Bras. Med. Trop. 2010, 43, 393–395. [Google Scholar] [CrossRef][Green Version]
- Yeshaw, Y.; Tsegaye, A.T.; Nigatu, S.G. Incidence of mortality and its predictors among adult visceral leishmaniasis patients at the university of gondar hospital: A retrospective cohort study. Infect. Drug Resist. 2020, 13, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Soeters, P.B.; Wolfe, R.R.; Shenkin, A. Hypoalbuminemia: Pathogenesis and Clinical Significance. JPEN. J. Parenter. Enteral Nutr. 2019, 43, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Daher, E.F.; Soares, D.S.; Filho, S.L.; Meneses, G.C.; Freitas, T.V.; Leite, T.T.; da Silva Junior, G.B. Hyponatremia and risk factors for death in human visceral leishmaniasis: New insights from a cross-sectional study in Brazil. BMC Infect. Dis. 2017, 17, 168. [Google Scholar] [CrossRef]
- Duarte, M.I.; da Matta, V.L.; Corbett, C.E.; Laurenti, M.D.; Chebabo, R.; Goto, H. Interstitial pneumonitis in human visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 1989, 83, 73–76. [Google Scholar] [CrossRef]
- Tuon, F.F.; Guedes, F.; Fernandes, E.R.; Pagliari, C.; Amato, V.S.; Seixas Duarte, M.I. In situ immune responses to interstitial pneumonitis in human visceral leishmaniasis. Parasite Immunol. 2009, 31, 98–103. [Google Scholar] [CrossRef]
- Bispo, A.J.B.; Almeida, M.L.D.; de Almeida, R.P.; Bispo Neto, J.; de Oliveira Brito, A.V.; França, C.M. Pulmonary involvement in human visceral leishmaniasis: Clinical and tomographic evaluation. PLoS ONE 2020, 15, e0228176. [Google Scholar] [CrossRef]
- Azadeh, N.; Limper, A.H.; Carmona, E.M.; Ryu, J.H. The Role of Infection in Interstitial Lung Diseases. Chest 2017, 152, 842–852. [Google Scholar] [CrossRef]
- Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6211. [Google Scholar] [CrossRef]
- Huo, R.; Guo, Q.; Hu, J.; Li, N.; Gao, R.; Mi, L.; Zhang, Z.; Liu, H.; Guo, Z.; Zhao, H.; et al. Therapeutic Potential of Janus Kinase Inhibitors for the Management of Interstitial Lung Disease. Drug Des. Devel. Ther. 2022, 16, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Libório, A.B.; Rocha, N.A.; Oliveira, M.J.C.; Franco, L.F.L.G.; Aguiar, G.B.R.; Pimentel, R.S.; Abreu, K.L.S.; Silva, G.B.; Daher, E.F. Acute Kidney Injury in Children with Visceral Leishmaniasis. Pediatr. Infect. Dis. J. 2012, 31, 451–454. [Google Scholar] [CrossRef]
- Oliveira, M.J.; Silva Júnior, G.B.; Abreu, K.L.; Rocha, N.A.; Garcia, A.V.; Franco, L.F.; Mota, R.M.; Libório, A.B.; Daher, E.F. Risk factors for acute kidney injury in visceral leishmaniasis (Kala-Azar). Am. J. Trop. Med. Hyg. 2010, 82, 449–453. [Google Scholar] [CrossRef] [PubMed]
- da Silva Junior, G.B.; Barros, E.J.G.; Daher, E.D.F. Kidney involvement in leishmaniasis—A review. Braz. J. Infect. Dis. 2013, 18, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Meneses, G.C.; De Francesco Daher, E.; da Silva Junior, G.B.; Bezerra, G.F.; da Rocha, T.P.; de Azevedo, I.E.P.; Libório, A.B.; Martins, A.M.C. Visceral leishmaniasis-associated nephropathy in hospitalised Brazilian patients: New insights based on kidney injury biomarkers. Trop. Med. Int. Health 2018, 23, 1046–1057. [Google Scholar] [CrossRef]
- Navarro, M.; Bonet, J.; Bonal, J.; Romero, R. Secondary amyloidosis with irreversible acute renal failure caused by visceral leishmaniasis in a patient with AIDS. Nefrologia 2006, 26, 745–746. [Google Scholar]
- Gangneux, J.-P.; Donaghy, L.; Marty, P. Liver involvement during visceral leishmaniasis. Gastroenterol. Clin. Biol. 2006, 30, 1027–1032. [Google Scholar] [CrossRef]
- Mathur, P.; Samantaray, J.C.; Samanta, P. High Prevalence of Functional Liver Derangement in Visceral Leishmaniasis at an Indian Tertiary Care Center. Clin. Gastroenterol. Hepatol. 2008, 6, 1170–1172. [Google Scholar] [CrossRef]
- Tawfik, A.K.; Amin, A.M.; Yousef, M.; El-Sayd, N.M.; Elashry, H.; Elkadeem, M.; Abd-Elsalam, S. IL-1α correlates with severity of hepatitis C virus-related liver diseases. J. Inflamm. Res. 2018, 11, 289–295. [Google Scholar] [CrossRef]
- Hall, P.; Cash, J. What is the real function of the liver “function” tests? Ulster Med. J. 2012, 81, 30–36. [Google Scholar]
- Alvarez-Nebreda, M.L.; Alvarez-Fernández, E.; Rada, S.; Brañas, F.; Marañón, E.; Vidán, M.T.; Serra-Rexach, J.A. Unusual duodenal presentation of leishmaniasis. J. Clin. Pathol. 2005, 58, 1321–1322. [Google Scholar] [CrossRef]
- Luz, J.G.G.; Naves, D.B.; de Carvalho, A.G.; Meira, G.A.; Dias, J.V.L.; Fontes, C.J.F.; de Carvalho, A.G.; Meira, G.A.; Dias, J.V.L.; Fontes, C.J.F. Visceral leishmaniasis in a Brazilian endemic area: An overview of occurrence, HIV coinfection and lethality. Rev. Inst. Med. Trop. Sao Paulo 2018, 60, e12. [Google Scholar] [CrossRef]
- Bhutta, Z.A.; Berkley, J.A.; Bandsma, R.H.J.; Kerac, M.; Trehan, I.; Briend, A. Severe childhood malnutrition. Nat. Rev. Dis. Prim. 2017, 3, 17067. [Google Scholar] [CrossRef]
- Baba, C.S.; Makharia, G.K.; Mathur, P.; Ray, R.; Gupta, S.D.; Samantaray, J.C. Chronic diarrhea and malabsorption caused by Leishmania donovani. Indian J. Gastroenterol. 2006, 25, 309–310. [Google Scholar]
- Boumis, E.; Chinello, P.; Della Rocca, C.; Paglia, M.G.; Proietti, M.F.; Petrosillo, N. Atypical disseminated leishmaniasis resembling post-kala-azar dermal leishmaniasis in an HIV-infected patient. Int. J. STD AIDS 2006, 17, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E. PKDL and other dermal lesions in HIV co-infected patients with Leishmaniasis: Review of clinical presentation in relation to immune responses. PLoS Negl. Trop. Dis. 2014, 8, e3258. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E. The immunology of post-kala-azar dermal leishmaniasis (PKDL). Parasit. Vectors 2016, 9, 464. [Google Scholar] [CrossRef] [PubMed]
- Goyal, V.; Das, V.N.R.; Singh, S.N.; Singh, R.S.; Pandey, K.; Verma, N.; Hightower, A.; Rijal, S.; Das, P.; Alvar, J.; et al. Long-term incidence of relapse and post-kala-azar dermal leishmaniasis after three different visceral leishmaniasis treatment regimens in Bihar, India. PLoS Negl. Trop. Dis. 2020, 14, e0008429. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K. Non-cirrhotic portal hypertension versus idiopathic portal hypertension. J. Gastroenterol. Hepatol. 2002, 17 (Suppl. S3), S204–S213. [Google Scholar] [CrossRef] [PubMed]
- Al-Jurayyan, N.A.M.; Al-Nasser, M.N.; Al-Fawaz, I.M.; Al Ayed, I.H.; Al Herbish, A.S.; M-Mazrou, A.M.; Al Sohaibani, M.O. The Haematological Manifestations of Visceral Leishmaniasis in Infancy and Childhood. J. Trop. Pediatr. 1995, 41, 143–148. [Google Scholar] [CrossRef]
- Rohrer, C.; Arni, U.; Deubelbeiss, K.A. Influence of the Spleen on the Distribution of Blood Neutrophils. Scand. J. Haematol. 2009, 30, 103–109. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef]
- Lawrence, S.M.; Corriden, R.; Nizet, V. How Neutrophils Meet Their End. Trends Immunol. 2020, 41, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Cotterell, S.E.J.; Engwerda, C.R.; Kaye, P.M. Enhanced Hematopoietic Activity Accompanies Parasite Expansion in the Spleen and Bone Marrow of Mice Infected with Leishmania donovani. Infect. Immun. 2000, 68, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Dixit, A.; Chatterjee, T.; Bhattacharya, M.; Bhattacharya, J.; Dutta, P.; Mahapatra, M.; Prasad Pati, H.; Choudhry, V.P.; Saxena, R.; et al. Disseminated intravascular coagulation as an unusual presentation of kala-azar: Report of two cases. Scand. J. Infect. Dis. 2004, 36, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Lomtadze, M.L.; Khochava, M.A.; Shalamberidze, I.A.; Kharaishvili, V.I.; Vorob’eva, E.O.; VI, K.; Vorob’eva, E.O. Study of intravascular coagulation activation markers in patients with visceral leishmaniasis. Georgian Med. News 2005, 124–125, 47–50. [Google Scholar]
- Thiagarajan, P.; Platelet Disorders Overview of Platelet Disorders. Medscape. Published 2021. Available online: https://emedicine.medscape.com/article/201722-overview#a2 (accessed on 23 October 2022).
- S Lima, S.; Cavalcante Braz, D.; Costa Silva, V.; J C Farias, T.; Zacarias, D.A.; da Silva, J.C.; Costa, C.H.N.; Costa, D.L. Biomarkers of the early response to treatment of visceral leishmaniasis: A prospective cohort study. Parasite Immunol. 2021, 43, e12797. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghazaly, J.; Al-Dubai, W. The clinical and biochemical characteristics of Yemeni adults and children with visceral leishmaniasis and the differences between them: A prospective cross-sectional study before and after treatment. Trop. Doct. 2016, 46, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.J.; Cheng, C.V.; Mattman, A.; Chen, L.Y.C. Polyclonal hypergammaglobulinaemia: Assessment, clinical interpretation, and management. Lancet Haematol. 2021, 8, e365–e375. [Google Scholar] [CrossRef]
- Zhao, E.J.; Carruthers, M.N.; Li, C.H.; Mattman, A.; Chen, L.Y.C. Conditions associated with polyclonal hypergammaglobulinemia in the IgG4-related disease era: A retrospective study from a hematology tertiary care center. Haematologica 2020, 105, e121–e123. [Google Scholar] [CrossRef]
- da Silva, P.E.F.; dos Santos Fonseca Junior, G.; Ambrozio, R.B.; Costa, M.G.S.T.; Machado, G.B.; de Carvalho, S.F.G.; de Oliveira, E.J.; Jorge, D.C.; de Almeida Silva Teixeira, L. LeishCare®: A software designed for the management of individuals with leishmaniases. Am. J. Trop. Med. Hyg. 2020, 103, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Abongomera, C.; Ritmeijer, K.; Vogt, F.; Buyze, J.; Mekonnen, Z.; Admassu, H.; Colebunders, R.; Mohammed, R.; Lynen, L.; Diro, E.; et al. Development and external validation of a clinical prognostic score for death in visceral leishmaniasis patients in a high HIV co-infection burden area in Ethiopia. PLoS ONE 2017, 12, e0178996. [Google Scholar] [CrossRef]
- Singh, K.; Singh, R.; Parija, S.C.; Faridi, M.M.A.; Bhatta, N. Clinical and laboratory study of kala-azar in children in Nepal. J. Trop. Pediatr. 1999, 45, 95–97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barati, M.; Sharifi, I.; Daie Parizi, M.; Fasihi Harandi, M. Bacterial infections in children with visceral leishmaniasis: Observations made in Kerman province, southern Iran, between 1997 and 2007. Ann. Trop. Med. Parasitol. 2008, 102, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Abdinia, B.; Oliaei-Motlagh, M.; Teimouri-Dereshki, A. Pediatric visceral leishmaniasis in northwest of Iran. Medicine 2016, 95, e5261. [Google Scholar] [CrossRef]
- Griffin, G.; Shenoi, S.; Hughes, G.C. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101515. [Google Scholar] [CrossRef]
- Bertoli, A.; Greco, A.V.; Caputo, S.; Caradonna, P.; Grieco, A.; Laghi, V. Visceral leishmaniasis presenting with hypertrigliceridaemia. Lancet 1982, 320, 504–505. [Google Scholar] [CrossRef]
- Lal, C.S.; Verma, R.B.; Verma, N.; Siddiqui, N.A.; Rabidas, V.N.; Pandey, K.; Singh, D.; Kumar, S.; Paswan, R.K.; Kumari, A.; et al. Hypertriglyceridemia: A possible diagnostic marker of disease severity in visceral leishmaniasis. Infection 2016, 44, 39–45. [Google Scholar] [CrossRef]
- Brun, R. Antiparasitic. Clin. Microbiol. Infect. 2012, 18, 16–17. [Google Scholar] [CrossRef][Green Version]
- Mottaghipisheh, H.; Kalantar, K.; Amanati, A.; Shokripour, M.; Shahriari, M.; Zekavat, O.R.; Zareifar, S.; Karimi, M.; Haghpanah, S.; Bordbar, M. Comparison of the clinical features and outcome of children with hemophagocytic lymphohistiocytosis (HLH) secondary to visceral leishmaniasis and primary HLH: A single-center study. BMC Infect. Dis. 2021, 21, 732. [Google Scholar] [CrossRef]
- González, J.L.; Insa, F.; Novoa, C.; Pizarro, M. Intestinal amyloidosis in hamsters with visceral leishmaniasis. Br. J. Exp. Pathol. 1986, 67, 353–360. [Google Scholar] [PubMed]
- de Freitas, J.C.C.; Lima, A.L.; Nunes-Pinheiro, D.C.S. Spleen damage in a dog naturally infected by Leishmania infantum. Rev. Soc. Bras. Med. Trop. 2017, 50, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B.; Toh, C.H.; Hoots, W.K.; Wada, H.; Levi, M.; Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb. Haemost. 2001, 86, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Wada, H. Disseminated intravascular coagulation. Clin. Chim. Acta 2004, 344, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Umemura, Y.; Wada, H.; Levy, J.H. Roles of Coagulation Abnormalities and Microthrombosis in Sepsis: Pathophysiology, Diagnosis, and Treatment. Arch. Med. Res. 2021, 52, 788–797. [Google Scholar] [CrossRef]
- Iba, T.; Levi, M.; Levy, J.H. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 2020, 46, 089–095. [Google Scholar] [CrossRef]
- Levi, M. Disseminated Intravascular Coagulation (DIC). Medscape. Published. 2022. Available online: https://emedicine.medscape.com/article/199627-overview (accessed on 12 May 2023).
- Butenas, S.; Orfeo, T.; Mann, K.G. Tissue factor in coagulation: Which? Where? When? Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1989–1996. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, W.R.; Poterucha, J.J. Evaluation of Elevated Liver Enzymes. Clin. Liver Dis. 2012, 16, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, E.T.; Pavlidis, T.E. Pathophysiological consequences of obstructive jaundice and perioperative management. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Andrade, T.M.; Carvalho, E.M.; Rocha, H. Bacterial infections in patients with visceral leishmaniasis. J. Infect. Dis. 1990, 162, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Kadivar, M.R.; Kajbaf, T.Z.; Karimi, A.; Alborzi, A. Childhood visceral leishmaniasis complicated by bacterial infections. East. Mediterr. Health J. 2000, 6, 879–883. [Google Scholar]
- Bone, R.C. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome. Crit. Care Med. 1996, 24, 163–172. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Shubin, N.J.; Monaghan, S.F.; Ayala, A. Anti-inflammatory mechanisms of sepsis. Contrib. Microbiol. 2011, 17, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol. 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, J.; Chen, S.; Ying, M.; Chen, G.; Liu, L.; Lun, Z.; Li, H.; Huang, H.; Li, Q.; et al. Malnutrition affects cholesterol paradox in coronary artery disease: A 41,229 Chinese cohort study. Lipids Health Dis. 2021, 20, 36. [Google Scholar] [CrossRef]
- Venet, F.; Monneret, G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 2018, 14, 121–137. [Google Scholar] [CrossRef]
- Liu, D.; Huang, S.-Y.; Sun, J.-H.; Zhang, H.-C.; Cai, Q.-L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-induced immunosuppression: Mechanisms, diagnosis and current treatment options. Mil. Med. Res. 2022, 9, 56. [Google Scholar] [CrossRef]
- Heffernan, D.S.; Monaghan, S.F.; Thakkar, R.K.; Machan, J.T.; Cioffi, W.G.; Ayala, A. Failure to normalize lymphopenia following trauma is associated with increased mortality, independent of the leukocytosis pattern. Crit. Care 2012, 16, R12. [Google Scholar] [CrossRef]
- Drewry, A.M.; Samra, N.; Skrupky, L.P.; Fuller, B.M.; Compton, S.M.; Hotchkiss, R.S. Persistent Lymphopenia After Diagnosis of Sepsis Predicts Mortality. Shock 2014, 42, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhang, S.; Peng, G.; Tang, Y.; Yang, Y. ICU and Sepsis: Role of Myeloid and Lymphocyte Immune Cells. J. Oncol. 2022, 2022, 7340266. [Google Scholar] [CrossRef]
- Finfer, S.; Venkatesh, B.; Hotchkiss, R.S.; Sasson, S.C. Lymphopenia in sepsis—An acquired immunodeficiency? Immunol. Cell Biol. 2023, 101, 535–544. [Google Scholar] [CrossRef]
- Francois, B.; Jeannet, R.; Daix, T.; Walton, A.H.; Shotwell, M.S.; Unsinger, J.; Monneret, G.; Rimmelé, T.; Blood, T.; Morre, M.; et al. Interleukin-7 restores lymphocytes in septic shock: The IRIS-7 randomized clinical trial. JCI Insight 2018, 3, e98960. [Google Scholar] [CrossRef]
- Silva-Freitas, M.L.; Corrêa-Castro, G.; Cota, G.F.; Giacoia-Gripp, C.; Rabello, A.; Teixeira Dutra, J.; de Vasconcelos, Z.F.M.; Savino, W.; Da-Cruz, A.M.; Santos-Oliveira, J.R. Impaired Thymic Output Can Be Related to the Low Immune Reconstitution and T Cell Repertoire Disturbances in Relapsing Visceral Leishmaniasis Associated HIV/AIDS Patients. Front. Immunol. 2020, 11, 953. [Google Scholar] [CrossRef] [PubMed]
- Takele, Y.; Mulaw, T.; Adem, E.; Shaw, C.J.; Franssen, S.U.; Womersley, R.; Kaforou, M.; Taylor, G.P.; Levin, M.; Müller, I.; et al. Immunological factors, but not clinical features, predict visceral leishmaniasis relapse in patients co-infected with HIV. Cell Rep. Med. 2022, 3, 100487. [Google Scholar] [CrossRef] [PubMed]
- Potestio, M.; D’Agostino, P.; Romano, G.C.; Milano, S.; Ferlazzo, V.; Aquino, A.; Di Bella, G.; Caruso, R.; Gambino, G.; Vitale, G.; et al. CD4+ CCR5+ and CD4+ CCR3+ lymphocyte subset and monocyte apoptosis in patients with acute visceral leishmaniasis. Immunology 2004, 113, 260–268. [Google Scholar] [CrossRef] [PubMed]
- de Souza, T.L.; da Silva, A.V.A.; Pereira, L.d.O.R.; Figueiredo, F.B.; Mendes Junior, A.A.V.; Menezes, R.C.; Mendes-da-Cruz, D.A.; Boité, M.C.; Cupolillo, E.; Porrozzi, R.; et al. Pro-Cellular Exhaustion Markers are Associated with Splenic Microarchitecture Disorganization and Parasite Load in Dogs with Visceral Leishmaniasis. Sci. Rep. 2019, 9, 12962. [Google Scholar] [CrossRef]
- Rytter, M.J.H.; Kolte, L.; Briend, A.; Friis, H.; Christensen, V.B. The Immune System in Children with Malnutrition—A Systematic Review. PLoS ONE 2014, 9, e105017. [Google Scholar] [CrossRef]
- Pierrakos, C.; Velissaris, D.; Bisdorff, M.; Marshall, J.C.; Vincent, J.-L. Biomarkers of sepsis: Time for a reappraisal. Crit. Care 2020, 24, 287. [Google Scholar] [CrossRef]
- Ashwin, H.; Sadlova, J.; Vojtkova, B.; Becvar, T.; Lypaczewski, P.; Schwartz, E.; Greensted, E.; Van Bocxlaer, K.; Pasin, M.; Lipinski, K.S.; et al. Characterization of a new Leishmania major strain for use in a controlled human infection model. Nat. Commun. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Grzymajlo, K. The Game for Three: Salmonella–Host–Microbiota Interaction Models. Front. Microbiol. 2022, 13, 854112. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).