
HLA-II-Dependent Neuroimmune Changes in Group A Streptococcal Necrotizing Fasciitis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. In Vivo GAS Infections
2.3. Tissue Collection
2.4. Gene Expression Changes in the Spleen and Hippocampus by Quantitative Real-Time PCR
2.5. Histological Tissue Preparation and Immunostaining
2.6. Quantification of GFAP and Iba-1 Immunoreactivity
2.7. Generation of Heatmaps and Correlation Plots
2.8. Statistical Analysis
3. Results
3.1. CLN Attenuated Skin GAS Burden in HLA-II DR3 Mice
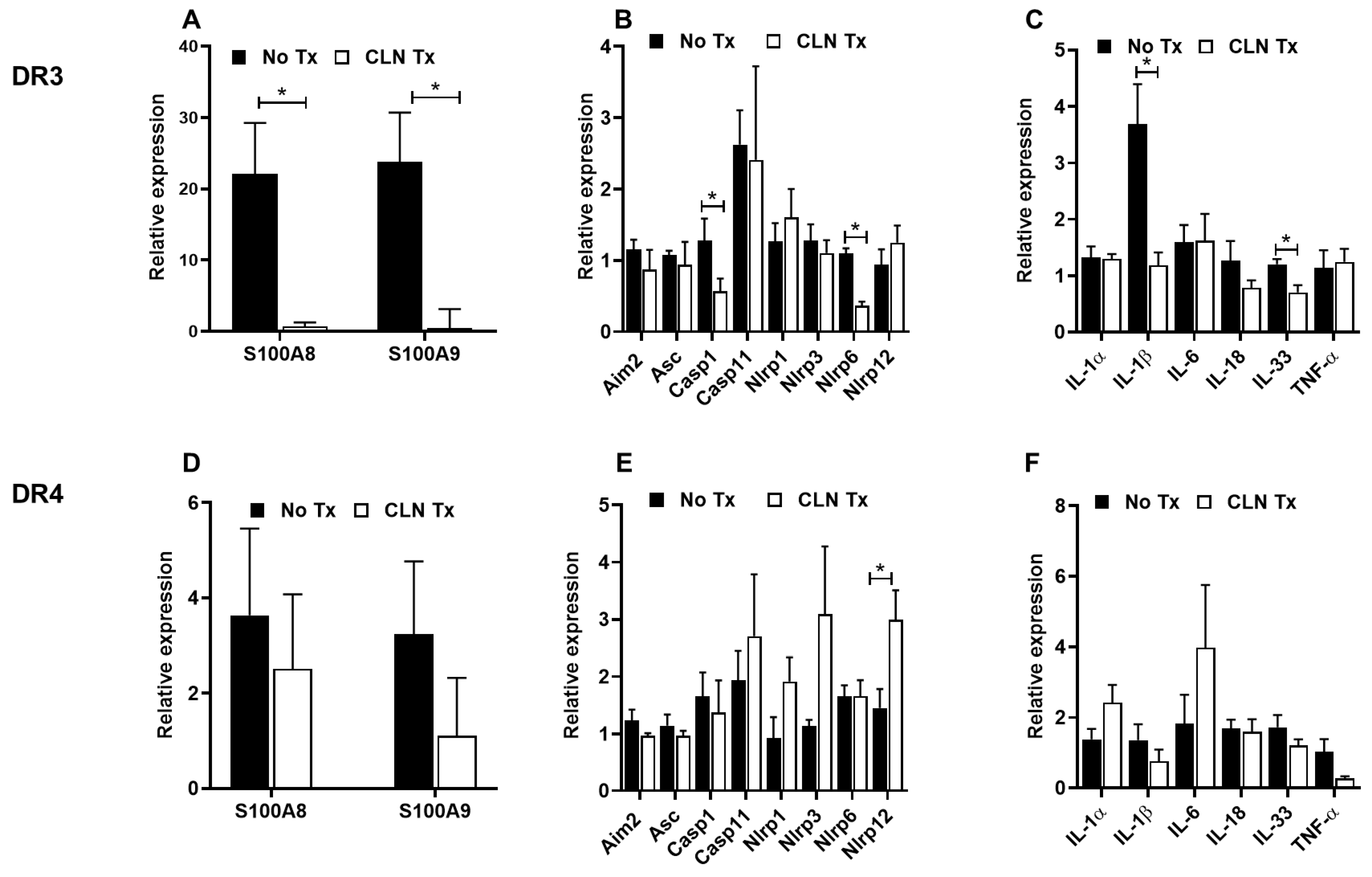
3.2. CLN Treatment Differentially Altered mRNA Levels of Numerous Genes in Spleens from GAS-Infected HLA-II DR3 Mice
3.3. CLN Treatment Differentially Altered mRNA Levels of Numerous Genes in Hippocampi from GAS-Infected HLA-II DR3 and DR4 Mice
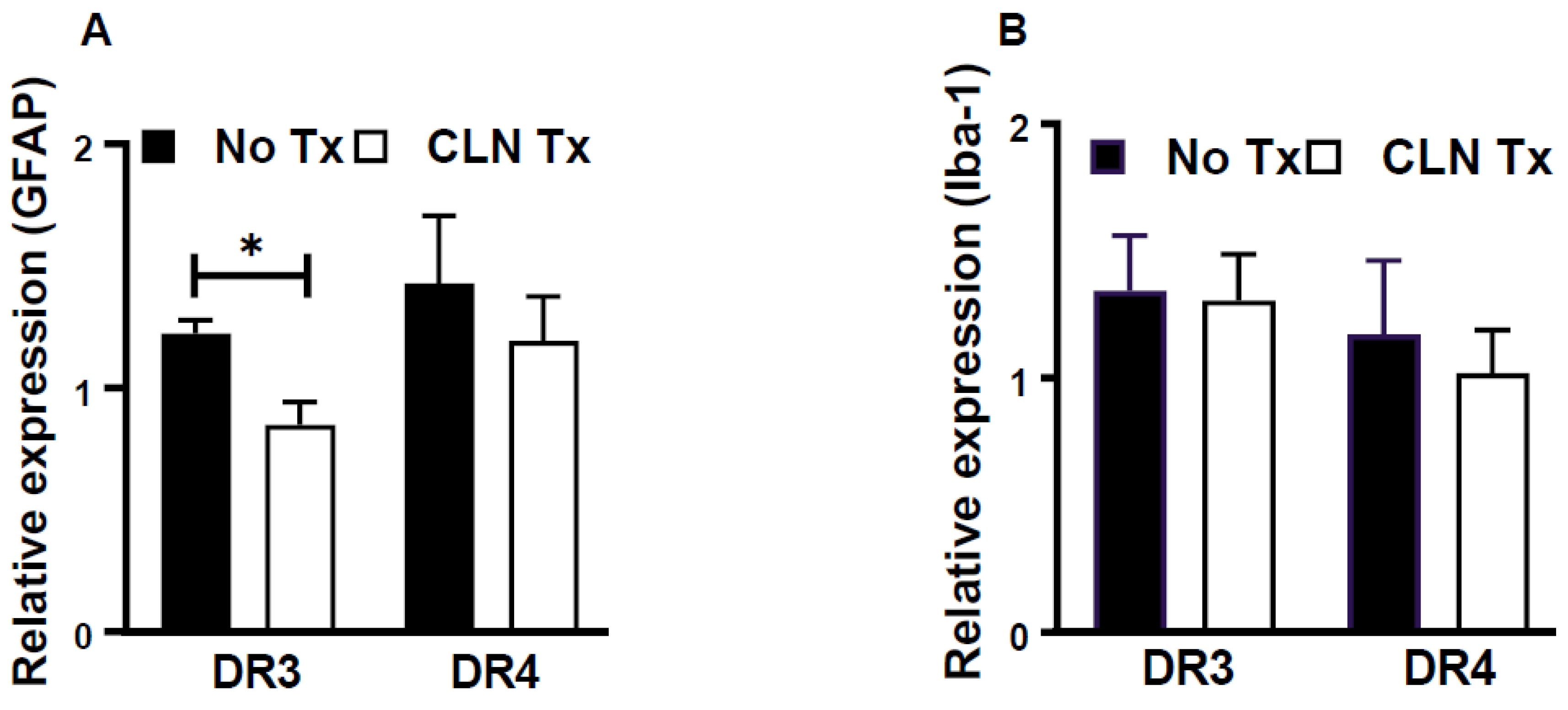
3.4. CLN Treatment Attenuated GFAP mRNA Levels in Hippocampi from GAS-Infected HLA-II DR3 Mice
3.5. Increased GFAP Immunoreactivity in Hippocampi of GAS-Infected HLA-II DR3 Mice
3.6. Analysis of mRNA Expression Patterns by Heatmap and Similarity Matrix Revealed Unique Clusters
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, M.J.; Barnett, T.C.; McArthur, J.D.; Cole, J.N.; Gillen, C.M.; Henningham, A.; Sriprakash, K.S.; Sanderson-Smith, M.L.; Nizet, V. Disease Manifestations and Pathogenic Mechanisms of Group A Streptococcus. Clin. Microbiol. Rev. 2014, 27, 264–301. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, F.; Zürcher, C.; Tarnutzer, A.; Schilcher, K.; Neff, A.; Keller, N.; Marques Maggio, E.; Poyart, C.; Schuepbach, R.A.; Zinkernagel, A.S.; et al. Clindamycin Affects Group a Streptococcus Virulence Factors and Improves Clinical Outcome. J. Infect. Dis. 2017, 215, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, D.B.; Schmitz, J.E.; Euler, C.W.; Fischetti, V.A. Novel Bacteriophage Lysin with Broad Lytic Activity Protects against Mixed Infection by Streptococcus Pyogenes and Methicillin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 2013, 57, 2743–2750. [Google Scholar] [CrossRef]
- Passàli, D.; Lauriello, M.; Passàli, G.C.; Passàli, F.M.; Bellussi, L. Group A Streptococcus and Its Antibiotic Resistance. Acta Otorhinolaryngol. Ital. 2007, 27, 27–32. [Google Scholar]
- Chandnani, H.K.; Jain, R.; Patamasucon, P. Group C Streptococcus Causing Rheumatic Heart Disease in a Child. J. Emerg. Med. 2015, 49, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Dubost, J.J.; Soubrier, M.; De Champs, C.; Ristori, J.M.; Sauvezie, B. Streptococcal Septic Arthritis in Adults. A Study of 55 Cases with a Literature Review. Jt. Bone Spine 2004, 71, 303–311. [Google Scholar] [CrossRef]
- Sims Sanyahumbi, A.; Colquhoun, S.; Wyber, R.; Carapetis, J.R. Global Disease Burden of Group A Streptococcus. Streptococcus Pyogenes Basic Biol. Clin. Manif. 2016, 5, 685–694. [Google Scholar] [CrossRef]
- Kim, S.W.; Grant, J.E.; Kim, S.I.; Swanson, T.A.; Bernstein, G.A.; Jaszcz, W.B.; Williams, K.A.; Schlievert, P.M. A Possible Association of Recurrent Streptococcal Infections and Acute Onset of Obsessive-Compulsive Disorder. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 252–260. [Google Scholar] [CrossRef]
- Quagliariello, A.; Del Chierico, F.; Russo, A.; Reddel, S.; Conte, G.; Lopetuso, L.R.; Ianiro, G.; Dallapiccola, B.; Cardona, F.; Gasbarrini, A.; et al. Gut Microbiota Profiling and Gut-Brain Crosstalk in Children Affected by Pediatric Acute-Onset Neuropsychiatric Syndrome and Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections. Front. Microbiol. 2018, 9, 675. [Google Scholar] [CrossRef]
- Risavi, B.L.; Iszkula, E.; Yost, B. Sydenham’s Chorea. J. Emerg. Med. 2019, 56, e119–e121. [Google Scholar] [CrossRef]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.M.; Blümcke, I.; Sander, J.W.; Löscher, W. Infections, Inflammation and Epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Kotb, M.; Norrby-Teglund, A.; McGeer, A.; El-Sherbini, H.; Dorak, M.T.; Khurshid, A.; Green, K.; Peeples, J.; Wade, J.; Thomson, G.; et al. An Immunogenetic and Molecular Basis for Differences in Outcomes of Invasive Group A Streptococcal Infections. Nat. Med. 2002, 8, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Zager, A.; Andersen, M.L.; Lima, M.M.S.; Reksidler, A.B.; Machado, R.B.; Tufik, S. Modulation of Sickness Behavior by Sleep: The Role of Neurochemical and Neuroinflammatory Pathways in Mice. Eur. Neuropsychopharmacol. 2009, 19, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, U.; Christie, D. Tourette syndrome and tics. Curr. Paediatr. 2003, 13, 42–46. [Google Scholar] [CrossRef]
- Martino, D.; Defazio, G.; Giovannoni, G. The PANDAS Subgroup of Tic Disorders and Childhood-Onset Obsessive-Compulsive Disorder. J. Psychosom. Res. 2009, 67, 547–557. [Google Scholar] [CrossRef]
- Martino, D.; Chiarotti, F.; Buttiglione, M.; Cardona, F.; Creti, R.; Nardocci, N.; Orefici, G.; Veneselli, E.; Rizzo, R. The Relationship between Group A Streptococcal Infections and Tourette Syndrome: A Study on a Large Service-Based Cohort. Dev. Med. Child Neurol. 2011, 53, 951–957. [Google Scholar] [CrossRef]
- Murphy, T.K.; Petitto, J.M.; Voeller, K.K.; Goodman, W.K. Obsessive Compulsive Disorder: Is There an Association with Childhood Streptococcal Infections and Altered Immune Function? Semin. Clin. Neuropsychiatry 2001, 6, 266–276. [Google Scholar] [CrossRef]
- Paul, S.P.; Jerwood, S. Group A Streptococcal Septicemia, Meningitis and Cerebral Abscess: Case Report and Literature Review. Turk. J. Pediatr. 2012, 54, 180. [Google Scholar]
- Bryant, P.A.; Robins-Browne, R.; Carapetis, J.R.; Curtis, N. Some of the People, Some of the Time Susceptibility to Acute Rheumatic Fever. Circulation 2009, 119, 742–753. [Google Scholar] [CrossRef]
- Cunningham, M.W.; Cox, C.J. Autoimmunity against Dopamine Receptors in Neuropsychiatric and Movement Disorders: A Review of Sydenham Chorea and Beyond. Acta Physiol. 2016, 216, 90–100. [Google Scholar] [CrossRef]
- Stevens, D.L.; Bisno, A.L.; Chambers, H.F.; Dellinger, E.P.; Goldstein, E.J.C.; Gorbach, S.L.; Hirschmann, J.V.; Kaplan, S.L.; Montoya, J.G.; Wade, J.C. Executive Summary: Practice Guidelines for the Diagnosis and Management of Skin and Soft Tissue Infections: 2014 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2014, 59, e10–e52. [Google Scholar] [CrossRef] [PubMed]
- Mangalam, A.K.; Rajagopalan, G.; Taneja, V.; David, C.S. HLA Class II Transgenic Mice Mimic Human Inflammatory Diseases. Adv. Immunol. 2008, 97, 65–147. [Google Scholar] [PubMed]
- Kong, Y.C.M.; Flynn, J.C.; Banga, J.P.; David, C.S. Application of HLA Class II Transgenic Mice to Study Autoimmune Regulation. Thyroid 2007, 17, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Shahi, S.K.; Ali, S.; Jaime, C.M.; Guseva, N.V.; Mangalam, A.K. HLA Class II Polymorphisms Modulate Gut Microbiota and Experimental Autoimmune Encephalomyelitis Phenotype. ImmunoHorizons 2021, 5, 627–646. [Google Scholar] [CrossRef]
- Ribeiro, S.P.; Rosa, D.S.; Fonseca, S.G.; Mairena, E.C.; Postól, E.; Oliveira, S.C.; Guilherme, L.; Kalil, J.; Cunha-Neto, E. A Vaccine Encoding Conserved Promiscuous HIV CD4 Epitopes Induces Broad T Cell Responses in Mice Transgenic to Multiple Common HLA Class II Molecules. PLoS ONE 2010, 5, e11072. [Google Scholar] [CrossRef]
- Das, P.; Chapoval, S.; Howard, V.; David, C.S.; Golde, T.E. Immune Responses against Abeta1-42 in HLA Class II Transgenic Mice: Implications for Abeta1-42 Immune-Mediated Therapies. Neurobiol. Aging 2003, 24, 969–976. [Google Scholar] [CrossRef]
- Khare, M.; Rodriguez, M.; David, C.S. HLA Class II Transgenic Mice Authenticate Restriction of Myelin Oligodendrocyte Glycoprotein-Specific Immune Response Implicated in Multiple Sclerosis Pathogenesis. Int. Immunol. 2003, 15, 535–546. [Google Scholar]
- Nooh, M.M.; El-Gengehi, N.; Kansal, R.; David, C.S.; Kotb, M. HLA Transgenic Mice Provide Evidence for a Direct and Dominant Role of HLA Class II Variation in Modulating the Severity of Streptococcal Sepsis. J. Immunol. 2007, 178, 3076–3083. [Google Scholar] [CrossRef]
- Germundson, D.L.; Nookala, S.; Smith, N.A.; Warda, Y.; Nagamoto-Combs, K. HLA-II Alleles Influence Physical and Behavioral Responses to a Whey Allergen in a Transgenic Mouse Model of Cow’s Milk Allergy. Front. Allergy 2022, 3, 870513. [Google Scholar] [CrossRef]
- Shahi, S.K.; Ghimire, S.; Lehman, P.; Mangalam, A.K. Obesity Induced Gut Dysbiosis Contributes to Disease Severity in an Animal Model of Multiple Sclerosis. Front. Immunol. 2022, 13, 966417. [Google Scholar] [CrossRef]
- Chowdhary, V.R.; Dai, C.; Tilahun, A.Y.; Hanson, J.A.; Smart, M.K.; Grande, J.P.; Rajagopalan, G.; Fu, S.-M.; David, C.S. A Central Role for HLA-DR3 in Anti-Smith Antibody Responses and Glomerulonephritis in a Transgenic Mouse Model of Spontaneous Lupus. J. Immunol. 2015, 195, 4660–4667. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Karau, M.J.; Greenwood-Quaintance, K.E.; Tilahun, A.Y.; Krogman, A.; David, C.S.; Pritt, B.S.; Patel, R.; Rajagopalan, G. Superantigen-Producing Staphylococcus Aureus Elicits Systemic Immune Activation in a Murine Wound Colonization Model. Toxins 2015, 7, 5308–5319. [Google Scholar] [CrossRef]
- Guédez, Y.; Kotby, A.; El-Demellawy, M.; Galal, A.; Thomson, G.; Zaher, S.; Kassem, S.; Kotb, M. HLA Class II Associations with Rheumatic Heart Disease Are More Evident and Consistent among Clinically Homogeneous Patients. Circulation 1999, 99, 2784–2790. [Google Scholar] [CrossRef]
- Fugger, L.; Svejgaard, A. Association of MHC and Rheumatoid Arthritis HLA-DR4 and Rheumatoid Arthritis: Studies in Mice and Men. Arthritis Res. Ther. 2000, 2, 208–211. [Google Scholar] [CrossRef]
- Hill, J.A.; Bell, D.A.; Brintnell, W.; Yue, D.; Wehrli, B.; Jevnikar, A.M.; Lee, D.M.; Hueber, W.; Robinson, W.H.; Cairns, E. Arthritis Induced by Posttranslationally Modified (Citrullinated) Fibrinogen in DR4-IE Transgenic Mice. J. Exp. Med. 2008, 205, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Becart, S.; Whittington, K.B.; Prislovsky, A.; Rao, N.L.; Rosloniec, E.F. The Role of Posttranslational Modifications in Generating Neo-Epitopes That Bind to Rheumatoid Arthritis-Associated HLA-DR Alleles and Promote Autoimmune T Cell Responses. PLoS ONE 2021, 16, e0245541. [Google Scholar] [CrossRef]
- Pal, S.; Tifrea, D.F.; Zhong, G.; de la Maza, L.M. Transcervical Inoculation with Chlamydia Trachomatis Induces Infertility in HLA-DR4 Transgenic and Wild-Type Mice. Infect. Immun. 2018, 86, e00722-17. [Google Scholar] [CrossRef] [PubMed]
- Hurtgen, B.J.; Castro-Lopez, N.; Jiménez-Alzate, M.D.P.; Cole, G.T.; Hung, C.-Y. Preclinical Identification of Vaccine Induced Protective Correlates in Human Leukocyte Antigen Expressing Transgenic Mice Infected with Coccidioides Posadasii. Vaccine 2016, 34, 5336–5343. [Google Scholar] [CrossRef]
- Hurtgen, B.J.; Hung, C.Y. Rational Design of T Lymphocyte Epitope-Based Vaccines against Coccidioides Infection. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017. [Google Scholar]
- Norrby-Teglund, A.; Nepom, G.T.; Kotb, M. Differential Presentation of Group A Streptococcal Superantigens by HLA Class II DQ and DR Alleles. Eur. J. Immunol. 2002, 32, 2570–2577. [Google Scholar] [CrossRef]
- Stanevicha, V.; Eglite, J.; Zavadska, D.; Sochnevs, A.; Shantere, R.; Gardovska, D. HLA Class II DR and DQ Genotypes and Haplotypes Associated with Rheumatic Fever among a Clinically Homogeneous Patient Population of Latvian Children. Arthritis Res. Ther. 2007, 9, R58. [Google Scholar] [CrossRef]
- Haydardedeoǧlu, F.E.; Tutkak, H.; Köse, K.; Düzgün, N. Genetic Susceptibility to Rheumatic Heart Disease and Streptococcal Pharyngitis: Association with HLA-DR Alleles. Tissue Antigens 2006, 68, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Drescher, K.M.; Geluk, A.; Bradley, D.S.; Rodriguez, M.; David, C.S. Complementation between Specific HLA-DR and HLA-DQ Genes in Transgenic Mice Determines Susceptibility to Experimental Autoimmune Encephalomyelitis. Hum. Immunol. 2000, 61, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Siemens, N.; Chakrakodi, B.; Shambat, S.M.; Morgan, M.; Bergsten, H.; Hyldegaard, O.; Skrede, S.; Arnell, P.; Madsen, M.B.; Johansson, L.; et al. Biofilm in Group: A Streptococcal Necrotizing Soft Tissue Infections. JCI Insight 2016, 1, e87882. [Google Scholar] [CrossRef] [PubMed]
- Emgård, J.; Bergsten, H.; McCormick, J.K.; Barrantes, I.; Skrede, S.; Sandberg, J.K.; Norrby-Teglund, A. MAIT Cells Are Major Contributors to the Cytokine Response in Group A Streptococcal Toxic Shock Syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 25923–25931. [Google Scholar] [CrossRef]
- Chella Krishnan, K.; Mukundan, S.; Alagarsamy, J.; Hur, J.; Nookala, S.; Siemens, N.; Svensson, M.; Hyldegaard, O.; Norrby-Teglund, A.; Kotb, M. Genetic Architecture of Group A Streptococcal Necrotizing Soft Tissue Infections in the Mouse. PLoS Pathog. 2016, 12, e1005732. [Google Scholar] [CrossRef]
- Nookala, S.; Mukundan, S.; Fife, A.; Alagarsamy, J.; Kotb, M. Heterogeneity in FoxP3- and GARP/LAP-Expressing T Regulatory Cells in an HLA Class II Transgenic Murine Model of Necrotizing Soft Tissue Infections by Group A Streptococcus. Infect. Immun. 2018, 86, 10–1128. [Google Scholar] [CrossRef]
- Krishnan, K.C.; Mukundan, S.; Alagarsamy, J.; Laturnus, D.; Kotb, M. Host Genetic Variations and Sex Differences Potentiate Predisposition, Severity, and Outcomes of Group A Streptococcus-Mediated Necrotizing Soft Tissue Infections. Infect. Immun. 2016, 84, 416–424. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; Manocha, G.D.; Puig, K.; Combs, C.K. An Improved Approach to Align and Embed Multiple Brain Samples in a Gelatin-Based Matrix for Simultaneous Histological Processing. J. Neurosci. Methods 2016, 261, 155–160. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Morriss, N.J.; Conley, G.M.; Ospina, S.M.; Meehan, W.P.; Qiu, J.; Mannix, R. Automated Quantification of Immunohistochemical Staining of Large Animal Brain Tissue Using QuPath Software. Neuroscience 2020, 429, 235–244. [Google Scholar] [CrossRef]
- Mannix, R.; Morriss, N.J.; Conley, G.M.; Meehan, W.P.; Nedder, A.; Qiu, J.; Float, J.; DiCesare, C.A.; Myer, G.D. Internal Jugular Vein Compression Collar Mitigates Histopathological Alterations after Closed Head Rotational Head Impact in Swine: A Pilot Study. Neuroscience 2020, 437, 132–144. [Google Scholar] [CrossRef]
- Lee, A.; Jiang, Z.; Zhu, L.; Ladiges, W. QuPath. A New Digital Imaging Tool for Geropathology. Aging Pathobiol. Ther. 2020, 2, 114–116. [Google Scholar] [CrossRef]
- Sahu, B.; Johnson, L.M.; Sohrabi, M.; Usatii, A.A.; Craig, R.M.J.; Kaelberer, J.B.; Chandrasekaran, S.P.; Kaur, H.; Nookala, S.; Combs, C.K. Effects of Probiotics on Colitis-Induced Exacerbation of Alzheimer’s Disease in AppNL-G-F Mice. Int. J. Mol. Sci. 2023, 24, 11551. [Google Scholar] [CrossRef]
- Simard, J.C.; Cesaro, A.; Chapeton-Montes, J.; Tardif, M.; Antoine, F.; Girard, D.; Tessier, P.A. S100A8 and S100A9 Induce Cytokine Expression and Regulate the NLRP3 Inflammasome via ROS-Dependent Activation of NF-ΚB1. PLoS ONE 2013, 8, e72138. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Wu, P.H.; Kang, C.C.; Tsai, Y.S.; Chou, C.K.; Liang, C.T.; Wu, J.J.; Tsai, P.J. Group A Streptococcus Subcutaneous Infection-Induced Central Nervous System Inflammation Is Attenuated by Blocking Peripheral TNF. Front. Microbiol. 2019, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Dileepan, T.; Smith, E.D.; Knowland, D.; Hsu, M.; Platt, M.; Bittner-Eddy, P.; Cohen, B.; Southern, P.; Latimer, E.; Harley, E.; et al. Group A Streptococcus Intranasal Infection Promotes CNS Infiltration by Streptococcal-Specific Th17 Cells. J. Clin. Investig. 2016, 126, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.; Thomas, S.; Williams, C. Clindamycin in the Treatment of Group G β-Haemolytic Streptococcal Infections. J. Infect. 2005, 51, e207–e211. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Gibbons, A.E.; Bergstrom, R.; Winn, V. The Eagle Effect Revisited: Efficacy of Clindamycin, Erythromycin, and Penicillin in the Treatment of Streptococcal Myositis. J. Infect. Dis. 1988, 158, 23–28. [Google Scholar] [CrossRef]
- Bryant, A.E.; Bayer, C.R.; Aldape, M.J.; McIndoo, E.; Stevens, D.L. Emerging Erythromycin and Clindamycin Resistance in Group A Streptococci: Efficacy of Linezolid and Tedizolid in Experimental Necrotizing Infection. J. Glob. Antimicrob. Resist. 2020, 22, 601–607. [Google Scholar] [CrossRef]
- Wang, S.S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.S.; Ma, J. S100A8-A9_rev_FrontImmu2018. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Aras, R.; Barron, A.M.; Pike, C.J. Caspase Activation Contributes to Astrogliosis. Brain Res. 2012, 1450, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Barclay, W.E.; Elizabeth Deerhake, M.; Inoue, M.; Nonaka, T.; Nozaki, K.; Luzum, A.; Aggarwal, N.; Miao, E.A.; Shinohara, M.L. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight 2022, 7, e155563. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, S.; Hu, Y.; Ma, Y.; Wu, Y.; Wu, C.; Liu, X.; Wang, B.; Hu, G.; Zhou, J.; et al. AIM2 Controls Microglial Inflammation to Prevent Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2021, 218, e20201796. [Google Scholar] [CrossRef]
- Ciążyńska, M.; Bednarski, I.A.; Wódz, K.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 Inflammasomes as a New Approach to Skin Carcinogenesis (Review). Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, G.; Zhong, B.; Zhang, H.; Ding, S.; Sun, Z.; Zhang, Y.; Li, W. NLRP1 Inflammasome Involves in Learning and Memory Impairments and Neuronal Damages during Aging Process in Mice. Behav. Brain Funct. 2021, 17, 11. [Google Scholar] [CrossRef]
- Tuncer, S.; Fiorillo, M.T.; Sorrentino, R. The Multifaceted Nature of NLRP12. J. Leukoc. Biol. 2014, 96, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Winglee, K.; Gharaibeh, R.Z.; Gauthier, J.; He, Z.; Tripathi, P.; Avram, D.; Bruner, S.; Fodor, A.; Jobin, C. Microbiota-Derived Metabolic Factors Reduce Campylobacteriosis in Mice. Gastroenterology 2018, 154, 1751–1763. [Google Scholar] [CrossRef]
- Best, E.L.; Freeman, J.; Wilcox, M.H. Models for the Study of Clostridium Difficile Infection. Gut Microbes 2012, 3, 145–167. [Google Scholar] [CrossRef]
- Nooh, M.M.; Nookala, S.; Kansal, R.; Kotb, M. Individual Genetic Variations Directly Effect Polarization of Cytokine Responses to Superantigens Associated with Streptococcal Sepsis: Implications for Customized Patient Care. J. Immunol. 2011, 186, 3156–3163. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (Forward) (5′-3′) | Primer Sequence (Reverse) (5′-3′) |
|---|---|---|
| S100A8 | GAGAAGGCCTTGAGCAACCTCATTG | CCTTGTGGCTGTCTTTGTGAGATG |
| S100A9 | GCAAGAAGATGGCCAACAAAGCAC | TCAAAGCTCAGCTGATTGTCCTGG |
| IL-1α | AAGACAAGCCTGTGTTGCTGAAGG | TCCCAGAAGAAAATGAGGTCGGTC |
| IL-1β | GCTTCAGGCAGGCAGTATC | AGGATGGGCTCTTCTTCAAAG |
| IL-6 | ACCGCTATGAAGTTCCTCTC | CTCTGTGAAGTCTCCTCTCC |
| IL-18 | ACCAAGTTCTCTTCGTTGAC | TCACAGCCAGTCCTCTTAC |
| IL-33 | CAATGACCAATCTGTTAGT | CATAGTAGCGTAGTAGCA |
| TNF-α | GGTTCTGTCCCTTTCACTCAC | TGCCTCTTCTGCCAGTTCC |
| AIM2 | ATAGGAGGAACAACAACAT | GCCATCTTCTGCTACATA |
| ASC | AGGAGTGGAGGGGAAAGC | AGAAGACGCAGGAAGATGG |
| CASP1 | AGGAATTCTGGAGCTTCAATCAG | TGGAAATGTGCCATCTTCTTT |
| CASP11 | GCTCTTACTTCATCACTA | AATATCTCGTCAAGGTTG |
| NLRP1 | GGTGTGCTGGTTGGTCTGC | GTGCTGTGGTGGTCTGTGAG |
| NLRP3 | GCTCCAACCATTCTCTGACC | AAGTAAGGCCGGAATTCACC |
| NLRP6 | GGACGAGAGGAAGGCAGAG | GCACACGAAGGGCACAAAG |
| NLRP12 | AAGAGATGAGATGCTACCTTGAGAG | ATGCCAACACTTCCTCCTTCAC |
| GFAP | GGTTGAATCGCTGGAGGAG | CTGTGAGGTCTGGCTTGG |
| Iba-1 | TTCCCAAGACCCACCTAG | TCCTCATACATCAGAATCATTC |
| RPLO | ACTGGTCTAGGACCCGAGAAG | TCCCACCTTGTCTCCAGTCT |
| RPL27 | ACATTGACGATGGCACCTC | GCTTGGCGATCTTCTTCTTG |
| GAPDH | TGTGTCCGTCGTGGATCTGA | CCTGCTTCACCACCTTCTTGA |
| β-Actin | AACCGTGAAAAGATGACCCAG | GCCTGGATGGCTACGGCTACGTACATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambigapathy, G.; Mukundan, S.; Nagamoto-Combs, K.; Combs, C.K.; Nookala, S. HLA-II-Dependent Neuroimmune Changes in Group A Streptococcal Necrotizing Fasciitis. Pathogens 2023, 12, 1000. https://doi.org/10.3390/pathogens12081000
Ambigapathy G, Mukundan S, Nagamoto-Combs K, Combs CK, Nookala S. HLA-II-Dependent Neuroimmune Changes in Group A Streptococcal Necrotizing Fasciitis. Pathogens. 2023; 12(8):1000. https://doi.org/10.3390/pathogens12081000
Chicago/Turabian StyleAmbigapathy, Ganesh, Santhosh Mukundan, Kumi Nagamoto-Combs, Colin K. Combs, and Suba Nookala. 2023. "HLA-II-Dependent Neuroimmune Changes in Group A Streptococcal Necrotizing Fasciitis" Pathogens 12, no. 8: 1000. https://doi.org/10.3390/pathogens12081000