
A New Paradigm in the Relationship between Gut Microbiota and Breast Cancer: β-glucuronidase Enzyme Identified as Potential Therapeutic Target

 ,
,  ,
,
Abstract
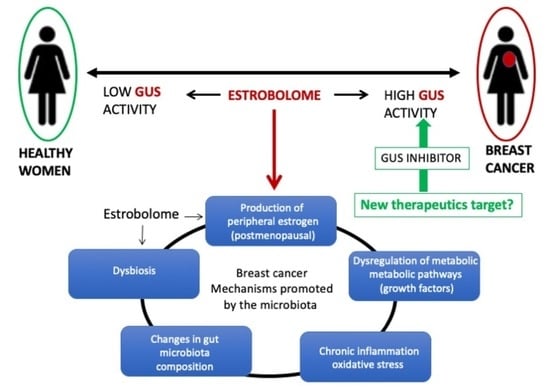
:
1. Introduction
2. Gut Microbiota, Diversity and Dysbiosis
3. Microbioma and Estrogen Metabolism: The Estrobolome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | Gene ID a | E Deconjugation b | PDB c Database (Accession ID) | Reference |
|---|---|---|---|---|---|
| Alistipes | EXC72_RS02090 ID: 78178623 | [25,32] | |||
| Akkermansia | muciniphila | GOZ73_RS09295 ID: 60881251 | [31,32] | ||
| Bacteroides | Fragilis | I6J55_RS13335 ID: 66330823 | Yes | 3CMG | [31,32] |
| cellulosilyticus | INE78_RS14030 ID: 66307762 | [32] | |||
| intestinalis | I1224_RS00440 ID: 69505108 | [32] | |||
| uniformis | INE75_RS18175 ID: 66283800 | 6NZG, 6D1N, 6D41, 6D50, 6D6W, 6D7F, 6D89, 6D8G | [32,33,34] | ||
| Ovatus | Bovatus_RS21525 ID: 29455654 | 6D8K | [32,34] | ||
| Dorei | FYB91_RS01050 ID: 56614211 | 6ED1 | [32,35] | ||
| massiliensis | I6J55_RS13335 ID: 66330823 | [32] | |||
| Vulgatus | GAIMETA21_RS00905 ID: 69838528 | [32] | |||
| Bacillus | thuringiensis | A9498_RS29930 ID: 39691567 | [32,36] | ||
| Bifidobacterium | Dentium | BIFDEN_RS03045 ID: 69535529 | 6LD0,6LD6, 6LDB, 6LDC, 6LDD | [32,37] | |
| Citrobacter | [32] | ||||
| Clostridium | perfringens | uidA [31] ID: 69447906 | yes | 6CXS, 6JKM, | [32,38,39] |
| Collinsella | tanakaei YIT 12063 | uidA ID: 62759750 | [32] | ||
| Dermabacter | [32] | ||||
| Edwardsiella | piscicida | uidA ID: 72529797 | [32] | ||
| Ictaluri | uidA ID: 69540280 | [32] | |||
| Escherichia | Coli | uidA ID: 946149 | yes | 6LEG, 3K46, 3K4A, 3K4D, 3LPF, 3LPG, 4JHZ, 5CZK, 6LEG, 6LEJ, 6LEL, 6LEM, 7PR6 | [31,32,37,39,40,41,42] |
| Eubacterium | Eligens | uidA ID: 41357285 | yes | 6BJW | [32,43] |
| Faecalibacterium | prausnitzii | uidA ID: 56863673 uidA ID: 34751772 | yes | 6U7I, 6ED2 | [32,35] |
| Fusicatenibacter | saccharivorans | 6NCY, 6NCZ | [32,44] | ||
| Lactobacillus | rhamnosus | RHM_0050 ID: 12473125 | yes | 6ECA | [32,35] |
| Gasseri | J3E66_RS04340 ID: 66468975 | [32,45] | |||
| Marvinbryantia | [32] | ||||
| Propionibacterium | Acnes | uidA ID: 12534223 | [32] | ||
| Parabacteroides | Merdae | DY317_RS05255 ID: 49202940 | 6DXU | [32] | |
| Johnsonii | HMPREF1077_RS04680 ID: 43351364 | [32] | |||
| Roseburia | Hominis | uidA ID: 77458459 | yes | 6MVH | [32] |
| intestinalis | uidA ID: 61434358 | [32] | |||
| Ruminococcus | Gnavus | N769_RS0107715 ID: 35896210 | yes | 6EC6 | [32,35] |
| Streptococcus | agalactiae | uidA ID: 66885601 | yes | 4JKL, 4JKK, 4JKL, | [32,39] |
| equisimilis | GGS_1280 ID: 13799427 | [32] | |||
| Tannerella | forsythia | BFO_RS10495 ID: 34759432 | [32,39] |
4. Axis Diet, Estrobolome and Breast Cancer
5. Other Activities of the Bacterial Estrobolome
6. Gut Microbiota β-glucuronidase Structure
7. Inhibitors of β-glucuronidase as Potential Anti-Cancer Treatment
8. Future Perspective
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory Today; IARC: Lyon, France, 2018.
- Horner, M.J.; Ries, L.A.G.; Krapcho, M.; Neyman, N.; Aminou, R.; Howlader, N.; Altekruse, S.F.; Feuer, E.J.; Huang, L. SEER Cancer Statistics Review; National Cancer Institute: Bethesda, MD, USA, 2015; pp. 1975–2012.
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early Breast Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Apostolou, P.; Fostira, F. Hereditary breast cancer: The era of new susceptibility genes. BioMed Res. Int. 2013, 2013, 747318. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Costa, D.A.; Nobre, J.G.; Batista, M.V.; Ribeiro, C.; Calle, C.; Cortes, A.; Marhold, M.; Negreiros, I.; Borralho, P.; Brito, M.; et al. Human Microbiota and Breast Cancer—Is There Any Relevant Link?—A Literature Review and New Horizons toward Personalised Medicine. Front. Microbiol. 2021, 12, 584332. [Google Scholar] [CrossRef]
- Roy, S.; Trinchieri, G. Microbiota: A Key Orchestrator of Cancer Therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The Resilience of the Intestinal Microbiota Influences Health and Disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A Guide from Sampling to Data Analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271. [Google Scholar] [CrossRef]
- Nogueira, A.R.; Shoenfeld, Y. Microbiome and Autoimmune Diseases: Cause and Effect Relationship. Curr. Opin. Rheumatol. 2019, 31, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Liao, M.; Yao, Z.; Liang, W.; Li, Q.; Liu, J.; Yang, H.; Ji, Y.; Wei, W.; Tan, A.; et al. Breast cancer in postmenopausal women is associated with an altered gut metagenome. Microbiome 2018, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Dzutsev, A.; Goldszmid, R.S.; Viaud, S.; Zitvogel, L.; Trinchieri, G. The Role of the Microbiota in Inflammation, Carcinogenesis, and Cancer Therapy. Eur. J. Immunol. 2015, 45, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the Gut Microbiota: An Unexpected Link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef]
- Mikó, E.; Kovács, T.; Sebő, É.; Tóth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabó, J.; Méhes, G.; Bai, P. Microbiome—Microbial Metabolome—Cancer Cell Interactions in Breast Cancer—Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal Interactions of the Intestinal Microbiota and Immune System. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef]
- Shapira, I.; Sultan, K.; Lee, A.; Taioli, E. Evolving Concepts: How Diet and the Intestinal Microbiome Act as Modulators of Breast Malignancy. ISRN Oncol. 2013, 2013, 693920. [Google Scholar] [CrossRef]
- Hullar, M.A.J.; Fu, B.C. Diet, the Gut Microbiome, and Epigenetics. Cancer J. 2014, 20, 170–175. [Google Scholar] [CrossRef]
- García-Castillo, V.; Sanhueza, E.; McNerney, E.; Onate, S.A.; García, A. Microbiota Dysbiosis: A New Piece in the Understanding of the Carcinogenesis Puzzle. J. Med. Microbiol. 2016, 65, 1347–1362. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and Malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor–Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Han, G.-Z.; Shim, J.-Y.; Wen, Y.; Jiang, X.-R. Quantitative Structure-Activity Relationship of Various Endogenous Estrogen Metabolites for Human Estrogen Receptor α and β Subtypes: Insights into the Structural Determinants Favoring a Differential Subtype Binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Mikó, E.; Ujlaki, G.; Sári, Z.; Bai, P. The Microbiome as a Component of the Tumor Microenvironment. In Tumor Microenvironment: Recent Advances; Springer: Berlin/Heidelberg, Germany, 2020; pp. 137–153. [Google Scholar]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and Transfer of Mitochondrial DNA via Exosomes Regulate Escape from Dormancy in Hormonal Therapy-Resistant Breast Cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Iván, J.; Major, E.; Sipos, A.; Kovács, K.; Horváth, D.; Tamás, I.; Bay, P.; Dombrádi, V.; Lontay, B. The Short-Chain Fatty Acid Propionate Inhibits Adipogenic Differentiation of Human Chorion-Derived Mesenchymal Stem Cells through the Free Fatty Acid Receptor 2. Stem Cells Dev. 2017, 26, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Jones, G.; Hua, X.; Xu, X.; Yu, G.; Flores, R.; Falk, R.T.; Gail, M.H.; Shi, J.; Ravel, J.; et al. Investigation of the Association Between the Fecal Microbiota and Breast Cancer in Postmenopausal Women: A Population-Based Case-Control Pilot Study. J. Natl. Cancer Inst. 2015, 107, djv147. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Pollet, R.M.; D’Agostino, E.H.; Walton, W.G.; Xu, Y.; Little, M.S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Creekmore, B.C.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977.e5. [Google Scholar] [CrossRef]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut Microbial β-Glucuronidases Reactivate Estrogens as Components of the Estrobolome That Reactivate Estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef]
- Jariwala, P.B.; Pellock, S.J.; Goldfarb, D.; Cloer, E.W.; Artola, M.; Simpson, J.B.; Bhatt, A.P.; Walton, W.G.; Roberts, L.R.; Major, M.B.; et al. Discovering the Microbial Enzymes Driving Drug Toxicity with Activity-Based Protein Profiling. ACS Chem. Biol. 2020, 15, 217–225. [Google Scholar] [CrossRef]
- Pellock, S.J.; Walton, W.G.; Biernat, K.A.; Torres-Rivera, D.; Creekmore, B.C.; Xu, Y.; Liu, J.; Tripathy, A.; Stewart, L.J.; Redinbo, M.R. Three Structurally and Functionally Distinct β-Glucuronidases from the Human Gut Microbe Bacteroides Uniformis. J. Biol. Chem. 2018, 293, 18559–18573. [Google Scholar] [CrossRef] [PubMed]
- Biernat, K.A.; Pellock, S.J.; Bhatt, A.P.; Bivins, M.M.; Walton, W.G.; Tran, B.N.T.; Wei, L.; Snider, M.C.; Cesmat, A.P.; Tripathy, A.; et al. Structure, Function, and Inhibition of Drug Reactivating Human Gut Microbial β-Glucuronidases. Sci. Rep. 2019, 9, 825. [Google Scholar] [CrossRef]
- Bang, W.Y.; Ban, O.-H.; Lee, B.S.; Oh, S.; Park, C.; Park, M.-K.; Jung, S.K.; Yang, J.; Jung, Y.H. Genomic-, Phenotypic-, and Toxicity-Based Safety Assessment and Probiotic Potency of Bacillus coagulans IDCC 1201 Isolated from Green Malt. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab026. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Chen, C.-Y.; Lin, T.-C.; Yeh, L.-F.; Hsieh, W.-C.; Gao, S.; Burnouf, P.-A.; Chen, B.-M.; Hsieh, T.-J.; Dashnyam, P.; et al. Entropy-Driven Binding of Gut Bacterial β-Glucuronidase Inhibitors Ameliorates Irinotecan-Induced Toxicity. Commun. Biol. 2021, 4, 280. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Pellock, S.J.; Biernat, K.A.; Walton, W.G.; Wallace, B.D.; Creekmore, B.C.; Letertre, M.M.; Swann, J.R.; Wilson, I.D.; Roques, J.R.; et al. Targeted Inhibition of Gut Bacterial β-Glucuronidase Activity Enhances Anticancer Drug Efficacy. Proc. Natl. Acad. Sci. USA 2020, 117, 7374–7381. [Google Scholar] [CrossRef]
- Wallace, B.D.; Roberts, A.B.; Pollet, R.M.; Ingle, J.D.; Biernat, K.A.; Pellock, S.J.; Venkatesh, M.K.; Guthrie, L.; O’Neal, S.K.; Robinson, S.J.; et al. Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol. 2015, 22, 1238–1249. [Google Scholar] [CrossRef]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.-A.; Mani, S.; et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Wallace, B.D.; Venkatesh, M.K.; Mani, S.; Redinbo, M.R. Molecular Insights into Microbial β-Glucuronidase Inhibition to Abrogate CPT-11 Toxicity. Mol. Pharmacol. 2013, 84, 208–217. [Google Scholar] [CrossRef]
- de Boer, C.; Armstrong, Z.; Lit, V.A.J.; Barash, U.; Ruijgrok, G.; Boyango, I.; Weitzenberg, M.M.; Schröder, S.P.; Sarris, A.J.C.; Meeuwenoord, N.J.; et al. Mechanism-Based Heparanase Inhibitors Reduce Cancer Metastasis In Vivo. Proc. Natl. Acad. Sci. USA 2022, 119, e2203167119. [Google Scholar] [CrossRef] [PubMed]
- Pellock, S.J.; Creekmore, B.C.; Walton, W.G.; Mehta, N.; Biernat, K.A.; Cesmat, A.P.; Ariyarathna, Y.; Dunn, Z.D.; Li, B.; Jin, J.; et al. Gut Microbial β-Glucuronidase Inhibition via Catalytic Cycle Interception. ACS Cent. Sci. 2018, 4, 868–879. [Google Scholar] [CrossRef]
- Pellock, S.J.; Walton, W.G.; Redinbo, M.R. Selecting a Single Stereocenter: The Molecular Nuances That Differentiate β-Hexuronidases in the Human Gut Microbiome. Biochemistry 2019, 58, 1311–1317. [Google Scholar] [CrossRef]
- Muccee, F.; Ghazanfar, S.; Ajmal, W.; Al-Zahrani, M. In-Silico Characterization of Estrogen Reactivating β-Glucuronidase Enzyme in GIT Associated Microbiota of Normal Human and Breast Cancer Patients. Genes 2022, 13, 1545. [Google Scholar] [CrossRef]
- Komorowski, A.S.; Pezo, R.C. Untapped “-Omics”: The Microbial Metagenome, Estrobolome, and Their Influence on the Development of Breast Cancer and Response to Treatment. Breast Cancer Res. Treat. 2020, 179, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [PubMed]
- Teng, N.M.Y.; Price, C.A.; McKee, A.M.; Hall, L.J.; Robinson, S.D. Exploring the Impact of Gut Microbiota and Diet on Breast Cancer Risk and Progression. Int. J. Cancer 2021, 149, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Bodai, B.I.; Nakata, T.E.; Wong, W.T.; Clark, D.R.; Lawenda, S.; Tsou, C.; Liu, R.; Shiue, L.; Cooper, N.; Rehbein, M.; et al. Lifestyle Medicine: A Brief Review of Its Dramatic Impact on Health and Survival. Perm. J. 2018, 22, 17-025. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Dannenberg, A.J. Obesity and Breast Cancer Prognosis: Weight of the Evidence. J. Clin. Oncol. 2011, 29, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kruper, L.; Dieli-Conwright, C.M.; Mortimer, J.E. The Impact of Obesity on Breast Cancer Diagnosis and Treatment. Curr. Oncol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Greenwood, D.C.; Lee, D.H.; Kim, R.; Aune, D.; Ju, W.; Hu, F.B.; Giovannucci, E.L. Adult Weight Gain and Adiposity-Related Cancers: A Dose-Response Meta-Analysis of Prospective Observational Studies. J. Natl. Cancer Inst. 2015, 107, djv088. [Google Scholar] [CrossRef]
- Ewertz, M.; Jensen, M.-B.; Gunnarsdóttir, K.Á.; Højris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of Obesity on Prognosis After Early-Stage Breast Cancer. J. Clin. Oncol. 2011, 29, 25–31. [Google Scholar] [CrossRef]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef] [PubMed]
- Bergom, C.; Kelly, T.; Bedi, M.; Saeed, H.; Prior, P.; Currey, A.D.; Wilson, J.; White, J. Does Size Matter: Examining the Association of BMI with Breast Cancer Recurrence and Survival in an Early Stage Breast Cancer Cohort with a High Median BMI. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, S47–S48. [Google Scholar] [CrossRef]
- Naaman, S.C.; Shen, S.; Zeytinoglu, M.; Iyengar, N.M. Obesity and Breast Cancer Risk: The Oncogenic Implications of Metabolic Dysregulation. J. Clin. Endocrinol. Metab. 2022, 107, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and Adverse Breast Cancer Risk and Outcome: Mechanistic Insights and Strategies for Intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef]
- Starek-Świechowicz, B.; Budziszewska, B.; Starek, A. Alcohol and Breast Cancer. Pharmacol. Rep. 2023, 75, 69–84. [Google Scholar] [CrossRef]
- Assi, N.; Rinaldi, S.; Viallon, V.; Dashti, S.G.; Dossus, L.; Fournier, A.; Cervenka, I.; Kvaskoff, M.; Turzanski-Fortner, R.; Bergmann, M.; et al. Mediation Analysis of the Alcohol-postmenopausal Breast Cancer Relationship by Sex Hormones in the EPIC Cohort. Int. J. Cancer 2020, 146, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.G.; Gonzalez-Reymundez, A.; Li, I.; Pathak, A.; Pathak, D.R.; de Los Campos, G.; Vazquez, A.I. Breast Cancer survival and the expression of genes related to alcohol drinking. PLoS ONE 2020, 15, e0228957. [Google Scholar] [CrossRef]
- Mühle, C.; Barry, B.; Weinland, C.; Kornhuber, J.; Lenz, B. Estrogen receptor 1 gene variants and estradiol activities in alcohol dependence. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 301–307. [Google Scholar] [CrossRef]
- Rachdaoui, N.; Sarkar, D.K. Effects of alcohol on the endocrine system. Endocrinol. Metab. Clin. N. Am. 2013, 42, 593–615. [Google Scholar] [CrossRef]
- Ho, A.M.; Geske, J.R.; Bakalkin, G.; Winham, S.J.; Karpyak, V.M. Correlations between sex-related hormones, alcohol dependence and alcohol craving. Drug Alcohol Depend. 2019, 197, 183–190. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, N.; Colditz, G.A. Links between Alcohol Consumption and Breast Cancer: A Look at the Evidence. Women’s Health 2015, 11, 65–77. [Google Scholar] [CrossRef]
- Freudenheim, J.L. Alcohol’s Effects on Breast Cancer in Women. Alcohol Res. 2020, 40, 11. [Google Scholar] [CrossRef]
- Malik, D.E.; David, R.M.; Gooderham, N.J. Ethanol potentiates the genotoxicity of the food-derived mammary carcinogen PhIP in human estrogen receptor-positive mammary cells: Mechanistic support for lifestyle factors (cooked red meat and ethanol) associated with mammary cancer. Arch. Toxicol. 2018, 92, 1639–1655. [Google Scholar] [CrossRef]
- Fan, S.; Meng, Q.; Gao, B.; Grossman, J.; Yadegari, M.; Goldberg, I.D.; Rosen, E.M. Alcohol Stimulates Estrogen Receptor Signaling in Human Breast Cancer Cell Lines. Cancer Res. 2000, 60, 5635–5639. [Google Scholar] [PubMed]
- Mutlu, E.; Keshavarzian, A.; Engen, P.; Forsyth, C.B.; Sikaroodi, M.; Gillevet, P. Intestinal Dysbiosis: A Possible Mechanism of Alcohol-Induced Endotoxemia and Alcoholic Steatohepatitis in Rats. Alcohol. Clin. Exp. Res. 2009, 33, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Abenavoli, L.; Cassano, V.; Rinninella, E.; Sorge, M.; Capretti, F.; Rasetti, C.; Baroni, G.S.; Luzza, F.; Santori, P.; et al. The Apparent Asymmetrical Relationship Between Small Bowel Bacterial Overgrowth, Endotoxemia, and Liver Steatosis and Fibrosis in Cirrhotic and Non-Cirrhotic Patients: A Single-Center Pilot Study. Front. Med. 2022, 9, 872428. [Google Scholar] [CrossRef]
- Zhu, B.T.; Bui, Q.D.; Weisz, J.; Liehr, J.G. Conversion of Estrone to 2- and 4-Hydroxyestrone by Hamster Kidney and Liver Microsomes: Implications for the Mechanism of Estrogen-Induced Carcinogenesis. Endocrinology 1994, 135, 1772–1779. [Google Scholar] [CrossRef]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and Improved Metabolic Health during a Dietary Intervention in Obesity: Relationship with Gut Microbiome Richness and Ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, S.; Kostopoulos, I.; de Vos, W.; Belzer, C. Akkermansia muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Morris, L.S.; Marchesi, J.R. The Gut Microbiome: The Role of a Virtual Organ in the Endocrinology of the Host. J. Endocrinol. 2013, 218, R37–R47. [Google Scholar] [CrossRef] [PubMed]
- Laborda-Illanes, A.; Sanchez-Alcoholado, L.; Dominguez-Recio, M.E.; Jimenez-Rodriguez, B.; Lavado, R.; Comino-Méndez, I.; Alba, E.; Queipo-Ortuño, M.I. Breast and Gut Microbiota Action Mechanisms in Breast Cancer Pathogenesis and Treatment. Cancers 2020, 12, 2465. [Google Scholar] [CrossRef]
- Klement, R.; Pazienza, V. Impact of Different Types of Diet on Gut Microbiota Profiles and Cancer Prevention and Treatment. Medicina 2019, 55, 84. [Google Scholar] [CrossRef] [PubMed]
- Ostan, R.; Lanzarini, C.; Pini, E.; Scurti, M.; Vianello, D.; Bertarelli, C.; Fabbri, C.; Izzi, M.; Palmas, G.; Biondi, F.; et al. Inflammaging and Cancer: A Challenge for the Mediterranean Diet. Nutrients 2015, 7, 2589–2621. [Google Scholar] [CrossRef] [PubMed]
- Caira, M.R.; Lonescu, C. Drug Metabolism: Current Concepts, 1st ed.; Springer: New York, NY, USA, 2010; Volume 7. [Google Scholar]
- Awolade, P.; Cele, N.; Kerru, N.; Gummidi, L.; Oluwakemi, E.; Singh, P. Therapeutic Significance of β-Glucuronidase Activity and Its Inhibitors: A Review. Eur. J. Med. Chem. 2020, 187, 111921. [Google Scholar] [CrossRef] [PubMed]
- ElRakaiby, M.; Dutilh, B.E.; Rizkallah, M.R.; Boleij, A.; Cole, J.N.; Aziz, R.K. Pharmacomicrobiomics: The Impact of Human Microbiome Variations on Systems Pharmacology and Personalized Therapeutics. OMICS 2014, 18, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the Drug-Microbiota Interactions in Anticancer Therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- de Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef]
- Parvez, M.M.; Basit, A.; Jariwala, P.B.; Gáborik, Z.; Kis, E.; Heyward, S.; Redinbo, M.R.; Prasad, B. Quantitative Investigation of Irinotecan Metabolism, Transport, and Gut Microbiome Activation. Drug Metab. Dispos. 2021, 49, 683–693. [Google Scholar] [CrossRef]
- Lin, X.B.; Dieleman, L.A.; Ketabi, A.; Bibova, I.; Sawyer, M.B.; Xue, H.; Field, C.J.; Baracos, V.E.; Gänzle, M.G. Irinotecan (CPT-11) Chemotherapy Alters Intestinal Microbiota in Tumour Bearing Rats. PLoS ONE 2012, 7, e39764. [Google Scholar] [CrossRef]
- Stringer, A.M.; Gibson, R.J.; Bowen, J.M.; Logan, R.M.; Ashton, K.; Yeoh, A.S.J.; Al-Dasooqi, N.; Keefe, D.M.K. Irinotecan-Induced Mucositis Manifesting as Diarrhoea Corresponds with an Amended Intestinal Flora and Mucin Profile. Int. J. Exp. Pathol. 2009, 90, 489–499. [Google Scholar] [CrossRef]
- Little, M.S.; Pellock, S.J.; Walton, W.G.; Tripathy, A.; Redinbo, M.R. Structural Basis for the Regulation of β-Glucuronidase Expression by Human Gut Enterobacteriaceae. Proc. Natl. Acad. Sci. USA 2018, 115, E152–E161. [Google Scholar] [CrossRef] [PubMed]
- LoGuidice, A.; Wallace, B.D.; Bendel, L.; Redinbo, M.R.; Boelsterli, U.A. Pharmacologic Targeting of Bacterial β-Glucuronidase Alleviates Nonsteroidal Anti-Inflammatory Drug-Induced Enteropathy in Mice. J. Pharmacol. Exp. Ther. 2012, 341, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Saitta, K.S.; Zhang, C.; Lee, K.K.; Fujimoto, K.; Redinbo, M.R.; Boelsterli, U.A. Bacterial β-Glucuronidase Inhibition Protects Mice against Enteropathy Induced by Indomethacin, Ketoprofen or Diclofenac: Mode of Action and Pharmacokinetics. Xenobiotica 2014, 44, 28–35. [Google Scholar] [CrossRef]
- An, J.; Kwon, H.; Lim, W.; Moon, B.-I. Staphylococcus Aureus-Derived Extracellular Vesicles Enhance the Efficacy of Endocrine Therapy in Breast Cancer Cells. J. Clin. Med. 2022, 11, 2030. [Google Scholar] [CrossRef]
- Donovitz, G.; Cotton, M. Breast Cancer Incidence Reduction in Women Treated with Subcutaneous Testosterone: Testosterone Therapy and Breast Cancer Incidence Study. Eur. J. Breast Health 2021, 17, 150–156. [Google Scholar] [CrossRef]
- Basit, A.; Amory, J.K.; Mettu, V.S.; Li, C.Y.; Heyward, S.; Jariwala, P.B.; Redinbo, M.R.; Prasad, B. Relevance of Human Aldoketoreductases and Microbial β-Glucuronidases in Testosterone Disposition. Drug Metab. Dispos. 2023, 51, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lacroix, C.; Wortmann, E.; Ruscheweyh, H.-J.; Sunagawa, S.; Sturla, S.J.; Schwab, C. Gut Microbial Beta-Glucuronidase and Glycerol/Diol Dehydratase Activity Contribute to Dietary Heterocyclic Amine Biotransformation. BMC Microbiol. 2019, 19, 99. [Google Scholar] [CrossRef] [PubMed]
- Landeka Jurčević, I.; Dora, M.; Guberović, I.; Petras, M.; Rimac Brnčić, S.; Đikić, D. Wine Lees Polyphenols as a Novel Functional Bioactive Compound in the Protection against Oxidative Stress and Hyperlipidemia. Food Technol. Biotechnol. 2017, 55, 109–116. [Google Scholar] [CrossRef]
- Miranda, A.; Steluti, J.; Fisberg, R.; Marchioni, D. Association between Coffee Consumption and Its Polyphenols with Cardiovascular Risk Factors: A Population-Based Study. Nutrients 2017, 9, 276. [Google Scholar] [CrossRef]
- Rothenberg, D.; Zhou, C.; Zhang, L. A Review on the Weight-Loss Effects of Oxidized Tea Polyphenols. Molecules 2018, 23, 1176. [Google Scholar] [CrossRef]
- Pérez-Jiménez, J.; Saura-Calixto, F. Macromolecular Antioxidants or Non-Extractable Polyphenols in Fruit and Vegetables: Intake in Four European Countries. Food Res. Int. 2015, 74, 315–323. [Google Scholar] [CrossRef]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of Dietary Compounds, Especially Polyphenols, with the Intestinal Microbiota: A Review. Eur. J. Nutr. 2015, 54, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Moga, M.; Dimienescu, O.; Arvatescu, C.; Mironescu, A.; Dracea, L.; Ples, L. The Role of Natural Polyphenols in the Prevention and Treatment of Cervical Cancer—An Overview. Molecules 2016, 21, 1055. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Bioactive Polyphenols and Cardiovascular Disease: Chemical Antagonists, Pharmacological Agents or Xenobiotics That Drive an Adaptive Response? Br. J. Pharmacol. 2017, 174, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.; Dean, O.; Turner, A.; Sureda, A.; Daglia, M.; Nabavi, S. Oxidative Stress and Post-Stroke Depression: Possible Therapeutic Role of Polyphenols? Curr. Med. Chem. 2014, 22, 343–351. [Google Scholar] [CrossRef]
- Leonidas, D.; Hayes, J.; Kato, A.; Skamnaki, V.; Chatzileontiadou, D.; Kantsadi, A.; Kyriakis, E.; Chetter, B.; Stravodimos, G. Phytogenic Polyphenols as Glycogen Phosphorylase Inhibitors: The Potential of Triterpenes and Flavonoids for Glycaemic Control in Type 2 Diabetes. Curr. Med. Chem. 2017, 24, 384–403. [Google Scholar] [CrossRef]
- Vitale, D.C.; Piazza, C.; Melilli, B.; Drago, F.; Salomone, S. Isoflavones: Estrogenic Activity, Biological Effect and Bioavailability. Eur. J. Drug Metab. Pharmacokinet. 2013, 38, 15–25. [Google Scholar] [CrossRef]
- Yokoyama, S.; Niwa, T.; Osawa, T.; Suzuki, T. Characterization of an O-Desmethylangolensin-Producing Bacterium Isolated from Human Feces. Arch. Microbiol. 2010, 192, 15–22. [Google Scholar] [CrossRef]
- Yokoyama, S.; Suzuki, T. Isolation and Characterization of a Novel Equol-Producing Bacterium from Human Feces. Biosci. Biotechnol. Biochem. 2008, 72, 2660–2666. [Google Scholar] [CrossRef]
- Matthies, A.; Blaut, M.; Braune, A. Isolation of a Human Intestinal Bacterium Capable of Daidzein and Genistein Conversion. Appl. Environ. Microbiol. 2009, 75, 1740–1744. [Google Scholar] [CrossRef]
- Raimondi, S.; Roncaglia, L.; De Lucia, M.; Amaretti, A.; Leonardi, A.; Pagnoni, U.M.; Rossi, M. Bioconversion of Soy Isoflavones Daidzin and Daidzein by Bifidobacterium Strains. Appl. Microbiol. Biotechnol. 2009, 81, 943–950. [Google Scholar] [CrossRef]
- Setchell, K.D.R.; Clerici, C. Equol: History, Chemistry, and Formation. J. Nutr. 2010, 140, 1355S–1362S. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.R.; Clerici, C. Equol: Pharmacokinetics and Biological Actions. J. Nutr. 2010, 140, 1363S–1368S. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.N.T.; Thybo, C.B.; Lykkeboe, S.; Rasmussen, L.M.; Frette, X.; Christensen, L.P.; Jeppesen, P.B. Combined bioavailable isoflavones and probiotics improve bone status and estrogen metabolism in postmenopausal osteopenic women: A randomized controlled trial. Am. J. Clin. Nutr. 2017, 106, 909–920. [Google Scholar] [CrossRef]
- Wang, J.; Xu, J.; Wang, B.; Shu, F.; Chen, K.; Mi, M. Equol promotes rat osteoblast proliferation and differentiation through activating estrogen receptor. Genet. Mol. Res. 2014, 13, 5055–5063. [Google Scholar] [CrossRef]
- Högger, P. Nutrition-Derived Bioactive Metabolites Produced by Gut Microbiota and Their Potential Impact on Human Health. Nutr. Med. 2013, 1, 122. [Google Scholar]
- Brunelli, E.; Pinton, G.; Chianale, F.; Graziani, A.; Appendino, G.; Moro, L. 8-Prenylnaringenin Inhibits Epidermal Growth Factor-Induced MCF-7 Breast Cancer Cell Proliferation by Targeting Phosphatidylinositol-3-OH Kinase Activity. J. Steroid Biochem. Mol. Biol. 2009, 113, 163–170. [Google Scholar] [CrossRef]
- Malik, P.; Singh, R.; Kumar, M.; Malik, A.; Mukherjee, T.K. Understanding the phytoestrogen genistein actions on breast cancer: Insights on estrogen receptor equivalence, pleiotropic essence and emerging paradigms in bioavailability modulation. Curr. Top. Med. Chem. 2023, 3, 1395–1413. [Google Scholar] [CrossRef]
- Wang, L.; Sun, R.; Zhang, Q.; Luo, Q.; Zeng, S.; Li, X.; Gong, X.; Li, Y.; Lu, L.; Hu, M.; et al. An Update on Polyphenol Disposition via Coupled Metabolic Pathways. Expert Opin. Drug Metab. Toxicol. 2019, 15, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Drendel, W.B.; Chen, Z.W.; Mathews, F.S.; Sly, W.S.; Grubb, J.H. Structure of Human Beta-Glucuronidase Reveals Candidate Lysosomal Targeting and Active-Site Motifs. Nat. Struct. Biol. 1996, 3, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Michikawa, M.; Ichinose, H.; Momma, M.; Biely, P.; Jongkees, S.; Yoshida, M.; Kotake, T.; Tsumuraya, Y.; Withers, S.G.; Fujimoto, Z.; et al. Structural and Biochemical Characterization of Glycoside Hydrolase Family 79 β-Glucuronidase from Acidobacterium Capsulatum. J. Biol. Chem. 2012, 287, 14069–14077. [Google Scholar] [CrossRef]
- Jayatilleke, K.M.; Hulett, M.D. Heparanase and the Hallmarks of Cancer. J. Transl. Med. 2020, 18, 453. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiang, J.; Jin, Y.; Kallemeijn, W.W.; Kuo, C.-L.; Artola, M.; Dai, W.; van Elk, C.; van Eijk, M.; van der Marel, G.A.; et al. Activity-Based Probes for Functional Interrogation of Retaining β-Glucuronidases. Nat. Chem. Biol. 2017, 13, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Jobin, C. The Struggle within: Microbial Influences on Colorectal Cancer. Inflamm. Bowel Dis. 2011, 17, 396–409. [Google Scholar] [CrossRef]
- Kong, R.; Liu, T.; Zhu, X.; Ahmad, S.; Williams, A.L.; Phan, A.T.; Zhao, H.; Scott, J.E.; Yeh, L.-A.; Wong, S.T.C. Old Drug New Use—Amoxapine and Its Metabolites as Potent Bacterial β-Glucuronidase Inhibitors for Alleviating Cancer Drug Toxicity. Clin. Cancer Res. 2014, 20, 3521–3530. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-W.; Tseng, C.-H.; Yang, C.-N.; Tzeng, C.-C.; Cheng, T.-C.; Leu, Y.-L.; Chuang, Y.-C.; Wang, J.-Y.; Lu, Y.-C.; Chen, Y.-L.; et al. Specific Inhibition of Bacterial β-Glucuronidase by Pyrazolo[4,3-c]Quinoline Derivatives via a PH-Dependent Manner To Suppress Chemotherapy-Induced Intestinal Toxicity. J. Med. Chem. 2017, 60, 9222–9238. [Google Scholar] [CrossRef]
- Letertre, M.P.M.; Bhatt, A.P.; Harvey, M.; Nicholson, J.K.; Wilson, I.D.; Redinbo, M.R.; Swann, J.R. Characterizing the Metabolic Effects of the Selective Inhibition of Gut Microbial β-Glucuronidases in Mice. Sci. Rep. 2022, 12, 17435. [Google Scholar] [CrossRef]




| Phylum (GUS Abundance %) | GUS Loop Classification | Localization | References |
|---|---|---|---|
| Bacteroidetes (52%) | L2 | Transported across the inner microbial membrane | [31,38,39] |
| mL1 | Periplasmic space | ||
| mL2 | Transported across the inner microbial membrane | ||
| NL | Periplasmic space | ||
| rare mL1,2 | Transported across the inner microbial membrane | ||
| Firmicutes (41%) | L1 | Intracellular | [31,38,39] |
| L2 | Transported across the inner microbial membrane | ||
| NL | Periplasmic space | ||
| mL1 | Periplasmic space | ||
| Verrucomicrobia (1.5%) | mL2 | Transported across the inner microbial membrane | [31,38,39] |
| Proteobacteria (4%) | L1 | Intracellular | [31,38,39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Murga, M.L.; Gil-Ortiz, F.; Serrano-García, L.; Llombart-Cussac, A. A New Paradigm in the Relationship between Gut Microbiota and Breast Cancer: β-glucuronidase Enzyme Identified as Potential Therapeutic Target. Pathogens 2023, 12, 1086. https://doi.org/10.3390/pathogens12091086
Fernández-Murga ML, Gil-Ortiz F, Serrano-García L, Llombart-Cussac A. A New Paradigm in the Relationship between Gut Microbiota and Breast Cancer: β-glucuronidase Enzyme Identified as Potential Therapeutic Target. Pathogens. 2023; 12(9):1086. https://doi.org/10.3390/pathogens12091086
Chicago/Turabian StyleFernández-Murga, M. Leonor, Fernando Gil-Ortiz, Lucía Serrano-García, and Antonio Llombart-Cussac. 2023. "A New Paradigm in the Relationship between Gut Microbiota and Breast Cancer: β-glucuronidase Enzyme Identified as Potential Therapeutic Target" Pathogens 12, no. 9: 1086. https://doi.org/10.3390/pathogens12091086
APA StyleFernández-Murga, M. L., Gil-Ortiz, F., Serrano-García, L., & Llombart-Cussac, A. (2023). A New Paradigm in the Relationship between Gut Microbiota and Breast Cancer: β-glucuronidase Enzyme Identified as Potential Therapeutic Target. Pathogens, 12(9), 1086. https://doi.org/10.3390/pathogens12091086