
Interferon-λ Activates a Differential Response in Peripheral Neurons That Is Effective against Alpha Herpesvirus Infections

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cells and Virus Strains
2.2. Primary Neuronal Culture
2.3. Viral Infection and Drug Treatment in Compartment Neuronal Cultures
2.4. Western Blot Analysis
2.5. Real-Time Quantitative PCR
2.6. Statistical Analysis
3. Results
3.1. IFN-λ Treatment Activates a Differential Response in Rat2 Cells versus Primary Neurons
3.2. IFN-λ Induces Differential Expression of Interferon Stimulated Genes (ISG) in Primary Neurons
3.3. Verification of Target ISG Induction in Rat2 Cells vs. Primary Neurons upon IFN-λ Treatment
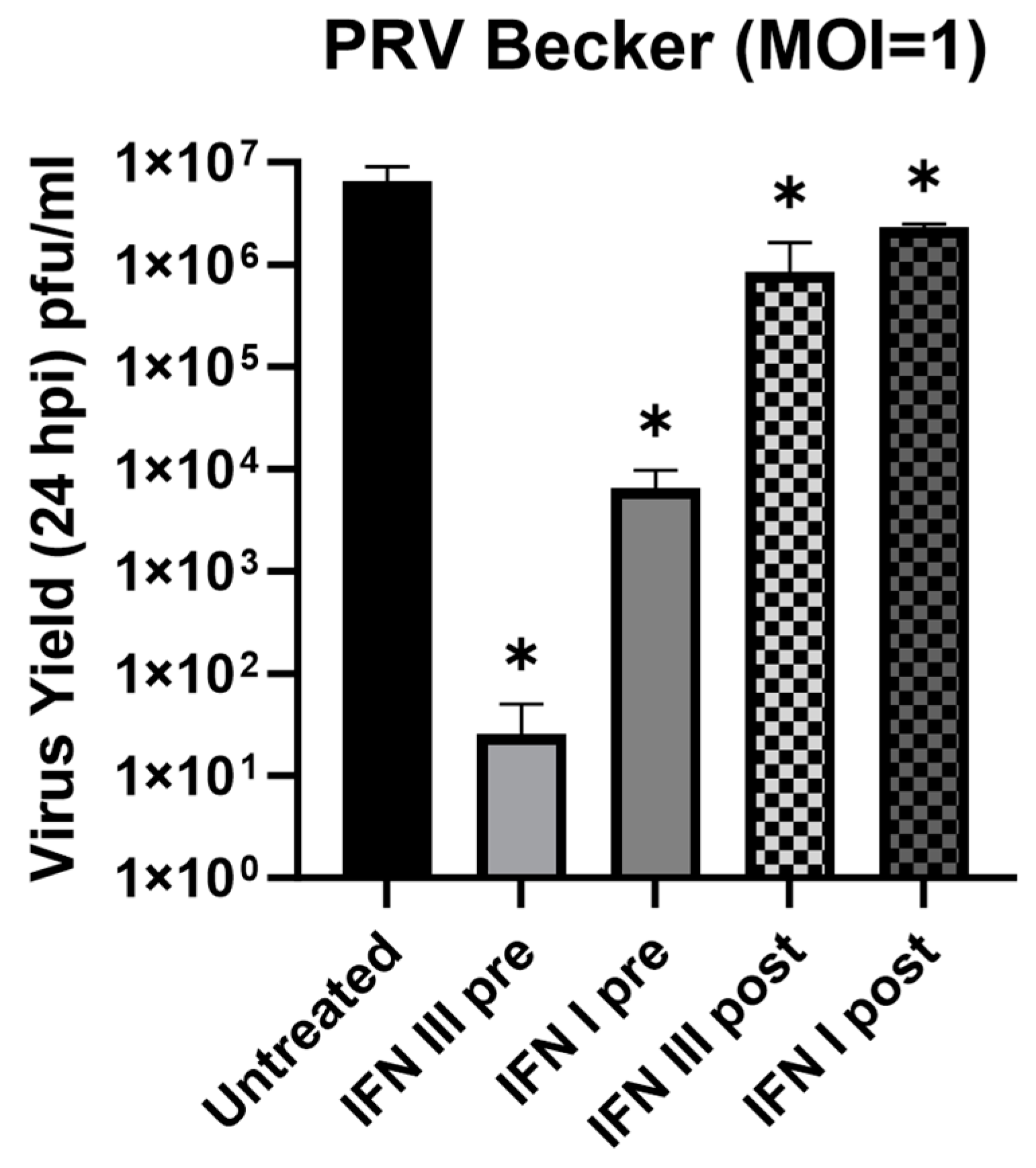
3.4. Antiviral Effect of IFN-λ in Rat2 Cells against Pseudorabies Virus (PRV)
3.5. Antiviral Effect of IFN-λ in Primary SCG Neurons against Pseudorabies Virus (PRV)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Broggi, A.; Granucci, F.; Zanoni, I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2019, 217, e20190295. [Google Scholar] [CrossRef]
- Menendez, C.M.; Carr, D.J. Defining nervous system susceptibility during acute and latent herpes simplex virus-1 infection. J. Neuroimmunol. 2017, 308, 43–49. [Google Scholar] [CrossRef]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef]
- Peng, T.; Zhu, J.; Phasouk, K.; Koelle, D.M.; Wald, A.; Corey, L. An effector phenotype of CD8+ T cells at the junction epithelium during clinical quiescence of herpes simplex virus 2 infection. J. Virol. 2012, 86, 10587–10596. [Google Scholar] [CrossRef] [PubMed]
- Sufiawati, I.; Tugizov, S.M. HIV-associated disruption of tight and adherens junctions of oral epithelial cells facilitates HSV-1 infection and spread. PLoS ONE 2014, 9, e88803. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus infections in the nervous system. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.C.; Sridhar, P.R.; Baldridge, M.T. Differential roles of interferons in innate responses to mucosal viral infections. Trends Immunol. 2021, 42, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef]
- Dellgren, C.; Gad, H.H.; Hamming, O.J.; Melchjorsen, J.; Hartmann, R. Human interferon-lambda3 is a potent member of the type III interferon family. Genes. Immun. 2009, 10, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mahlakoiv, T.; Hernandez, P.; Gronke, K.; Diefenbach, A.; Staeheli, P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015, 11, e1004782. [Google Scholar] [CrossRef]
- Lan, F.; Wane, X.D.; Nauwynck, H.; Holtappels, G.; Zhang, L.; Johnston, S.L.; Papadopoulos, N.G.; Bachert, C.; Zhang, N. Th2 biased upper airway inflammation is associated with an impaired response to viral infection with Herpes simplex virus 1. Rhinology 2016, 54, 141–149. [Google Scholar] [PubMed]
- Rodriguez-Hernandez, C.J.; Sokoloski, K.J.; Stocke, K.S.; Dukka, H.; Jin, S.; Metzler, M.A.; Zaitsev, K.; Shpak, B.; Shen, D.; Miller, D.P.; et al. Microbiome-mediated incapacitation of interferon lambda production in the oral mucosa. Proc. Natl. Acad. Sci. USA 2021, 118, e2105170118. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.T.; Kennedy, E.A.; Brigleb, P.H.; Taylor, G.M.; Urbanek, K.; Bricker, T.L.; Lee, S.; Shin, H.; Dermody, T.S.; Boon, A.C.M.; et al. Disruption of type III interferon (IFN) genes Ifnl2 and Ifnl3 recapitulates loss of the type III IFN receptor in the mucosal antiviral response. J. Virol. 2019, 93, e01073-19. [Google Scholar] [CrossRef]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda interferon (IFN-λ), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef]
- Peng, T.; Zhu, J.; Klock, A.; Phasouk, K.; Huang, M.L.; Koelle, D.M.; Wald, A.; Corey, L. Evasion of the mucosal innate immune system by herpes simplex virus type 2. J. Virol. 2009, 83, 12559–12568. [Google Scholar] [CrossRef]
- Rosato, P.C.; Leib, D.A. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 2015, 11, e1005028. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, S.; Zhou, L.; Ye, L.; Wang, X.; Ho, J.; Ho, W. Interferon lambda inhibits herpes simplex virus type I infection of human astrocytes and neurons. Glia 2011, 59, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Koyuncu, O.O.; Greco, T.M.; Diner, B.A.; Cristea, I.M.; Enquist, L.W. Two Modes of the Axonal Interferon Response Limit Alphaherpesvirus Neuroinvasion. mBio 2016, 7, e02145-15. [Google Scholar] [CrossRef] [PubMed]
- Platt, K.B.; Mare, C.J.; Hinz, P.N. Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky’s disease) virus: Thermal sensitivity and rabbit virulence markers. Arch. Virol. 1979, 60, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, T.; Ch’ng, T.H.; Flood, E.A.; Gross, S.P.; Enquist, L.W. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 2005, 79, 3903–3919. [Google Scholar] [CrossRef] [PubMed]
- Curanović, D.; Ch’ng, T.H.; Szpara, M.; Enquist, L. Compartmented neuron cultures for directional infection by alpha herpesviruses. Curr. Protoc. Cell Biol. 2009, 43, 26.4.1–26.4.23. [Google Scholar] [CrossRef]
- Hean Ch’ng, T.; Flood, E.A.; Enquist, L.W. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. DNA Viruses Methods Protoc. 2005, 292, 299–315. [Google Scholar]
- Cheung, A.K. Detection of pseudorabies virus transcripts in trigeminal ganglia of latently infected swine. J. Virol. 1989, 63, 2908–2913. [Google Scholar] [CrossRef]
- Jacobs, S.; Wavreil, F.; Schepens, B.; Gad, H.H.; Hartmann, R.; Rocha-Pereira, J.; Neyts, J.; Saelens, X.; Michiels, T. Species specificity of type III interferon activity and development of a sensitive luciferase-based bioassay for quantitation of mouse interferon-λ. J. Interferon Cytokine Res. 2018, 38, 469–479. [Google Scholar] [CrossRef]
- Edmans, J.; Clitherow, K.; Murdoch, C.; Hatton, P.; Spain, S.; Colley, H. Mucoadhesive Electrospun Fibre-Based Technologies for Oral Medicine. Pharmaceutics 2020, 12, 504. [Google Scholar] [CrossRef]
- Bolen, C.R.; Ding, S.; Robek, M.D.; Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Enquist, L.W. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 4873–4878. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Enquist, L.W.; Pomeranz, L.E. The alpha-herpesviruses: Molecular pathfinders in nervous system circuits. Trends Mol. Med. 2008, 14, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Paladino, P.; Mossman, K.L. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J. Interferon Cytokine Res. 2009, 29, 599–607. [Google Scholar] [CrossRef]
- Frade, J.M.; Ovejero-Benito, M.C. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle 2015, 14, 712–720. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef]
- Yin, Y.; Romero, N.; Favoreel, H.W. Pseudorabies virus inhibits type I and type III interferon-induced signaling via proteasomal degradation of Janus kinases. J. Virol. 2021, 95, e00793-21. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. Jak-Stat 2013, 2, e23931. [Google Scholar] [CrossRef]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune response in Stat2 knockout mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011, 7, e1001297. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef]
- Iversen, M.B.; Ank, N.; Melchjorsen, J.; Paludan, S.R. Expression of type III interferon (IFN) in the vaginal mucosa is mediated primarily by dendritic cells and displays stronger dependence on NF-κB than type I IFNs. J. Virol. 2010, 84, 4579–4586. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, T.A.; Bhushal, S.; Kalinke, U.; Wirth, D.; Hauser, H.; Köster, M.; Hornef, M.W. Identification of a predominantly interferon-λ-induced transcriptional profile in murine intestinal epithelial cells. Front. Immunol. 2017, 8, 1302. [Google Scholar] [CrossRef]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef]
- Dziennis, S.; Alkayed, N. Role of Signal Transducer and Activator of Transcription 3 in Neuronal Survival and Regeneration. Rev. Neurosci. 2008, 19, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef]
- Liu, T.; Xu, Z.Z.; Park, C.K.; Berta, T.; Ji, R.R. Toll-like receptor 7 mediates pruritus. Nat. Neurosci. 2010, 13, 1460–1462. [Google Scholar] [CrossRef]
- Zhou, L.; Li, J.L.; Zhou, Y.; Liu, J.B.; Zhuang, K.; Gao, J.F.; Liu, S.; Sang, M.; Wu, J.G.; Ho, W.Z. Induction of interferon-λ contributes to TLR3 and RIG-I activation-mediated inhibition of herpes simplex virus type 2 replication in human cervical epithelial cells. MHR Basic Sci. Reprod. Med. 2015, 21, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Song, R.; Greco, T.M.; Cristea, I.M.; Enquist, L.W. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. MBio 2015, 6, 10–1128. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Actb | CGTCCACCCGCGAGTACAACC | CGACGACGAGCGCAGCGATATCG |
| CCL2 | GCTAATGCATCCACTCTC | GTTTAACATTACTTAAGGC |
| Cd38 | GCGTAGTCTTCATTGGTGATG | CGCTGACATCATCTTGGGACGC |
| Cd69 | CGCAGTCTACAGAAGCAAC | GACTGAAACACTGGATTGGGC |
| Crp | GGGGCAGGAGCAGGACTCG | CATAGACTGCATTGATCTG |
| Cxcl10 | GGCTTCCCAATTCTCTAAGAGC | GAGCAGGCTGGGGCATGGC |
| Gapdh | GCAACAATGTCCACTTTGTC | GCCTGGTTACCAGGGCTGC |
| Ifit1 | CGCTACGCAGCCAAGTATTTCC | CTGTTGCTTGTAACAGAGCCC |
| Ifit2 | GCTGAGTGCTTTGACACAGC | GCTCTCCAATGATTCCTTAC |
| Ifit3 | GCAACTGCAGAATACAGG | CTTATTCATGTACTCCATAG |
| Ifitm3 | CCCCCAGTGGCAGCGTTCACG | GAACAGTCCGACAGAGCC |
| Ifna1 | GCCAGTAGGGAGTCTTCCTGG | GCTTGGGATGCAACCCTCC |
| Irf9 | CCAACCTGCCACTCTCGC | CCTTGCACTTTCTCTTGGCC |
| Isg15 | GGTCCCCTGAAACTAAGG | GCTGACCCAGTGAGTCTCTC |
| Jak1 | CGAAGGCAGAAACTCCATG | CGGAACCTCTACCACGAGAAC |
| Oas1a | CGAATGTATCTCCCTGGGG | GCATAGACCGTGAGCAGC |
| Oas1b | GCGCCAATTCAGGAAGTACAGC | CCCAACCCACAATTCTACG |
| Oas2 | GGTGATGATCGATGTGCTG | CTGGTCTTGGAACTGTTTG |
| Stat1 | GCATTTAAAGTCATATTCATC | CCGTGATGTTAGATAAAC |
| Stat2 | GGAGTGGGTGGGAAACGGG | GCAACATCTCCCACTGCGCC |
| Stat3 | GCAGGAATCGGCTATACTGC | CCTGGCGCCTTGGATTGAGAGC |
| Tlr7 | GGACTTCTTCAAGAATCTGC | GGCTAAGGAGAGAGTCGTTAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, S.; Luong, K.T.Y.; Nua, T.; Koyuncu, O.O. Interferon-λ Activates a Differential Response in Peripheral Neurons That Is Effective against Alpha Herpesvirus Infections. Pathogens 2023, 12, 1142. https://doi.org/10.3390/pathogens12091142
Salazar S, Luong KTY, Nua T, Koyuncu OO. Interferon-λ Activates a Differential Response in Peripheral Neurons That Is Effective against Alpha Herpesvirus Infections. Pathogens. 2023; 12(9):1142. https://doi.org/10.3390/pathogens12091142
Chicago/Turabian StyleSalazar, Stephanie, Khanh T. Y. Luong, Taulima Nua, and Orkide O. Koyuncu. 2023. "Interferon-λ Activates a Differential Response in Peripheral Neurons That Is Effective against Alpha Herpesvirus Infections" Pathogens 12, no. 9: 1142. https://doi.org/10.3390/pathogens12091142